要旨¶
X線および中性子小角散乱(SAXSおよびSANS)は、構造パラメータを抽出し、 溶液中の生体高分子、複合体および集合体の全体構造および形状を 決定するために使用される技術である。サンプルから測定された散乱強度は、 溶媒、緩衝液成分ならびに目的の生体高分子を含む照射された サンプル体積内のすべての原子からの寄与を含む。構造情報を得るためには、 サンプル溶液中の高分子からの正味の散乱を得るよう、すなわち、サンプル散乱から バックグラウンド散乱寄与を正確に差し引くことができるように、サンプルと 正確に一致した溶媒ブランクを調製することが不可欠である。
さらに、コンタミ、凝集物、ミスマッチした溶媒、放射線損傷または 他の要因によって引き起こされる試料の異種性は、データ解析に深刻な影響を与え、 複雑にする可能性があるため、測定期間中、試料は純粋で単分散であることが不可欠である。 この手順書は、SAXSとSANSの基本的な物理を概説し、背景となる散乱法の原理が どのようにして最終的に散乱実験のために十分に高品質なサンプルを調整するための技術的根拠 となっているかを明らかにする。この手順書は、ゲル電気泳動、 サイズ排除クロマトグラフィーおよび光散乱を用いて、SAXSおよびSANSの両方について タンパク質および核酸試料を調製および特徴付ける方法を記載する。また、X線 (in-lineサイズ排除クロマトグラフィーSAXS)および中性子に特有の手順、 具体的にはコントラストマッチング/バリエーション実験および タンパク質の重水素標識のためのサンプルの調製も含まれる。
序論¶
現代のシステム構造生物学は、相互接続された複雑な生体高分子ネットワーク、そして、これらのネットワークがどのようにして分子レベルのコミュニケーションに仲介し細胞応答に影響を与えるか、その解読に大きな課題を抱えています。X線結晶学、核磁気共鳴分光法(NMR)、そして最近では高分解能電子顕微鏡法(EM)のような高分解能構造決定方法は、タンパク質や他の生体高分子の原子レベルの構造詳細を明らかにするためには別格です。 しかし、高解像度技術を使用して異なるサンプル環境中の生体高分子、複合体および集合体の立体構造応答を評価することはますますむずかしくなっている。 装置およびソフトウェア開発の継続的な進歩とデータ収集、解析、モデリングのための自動化が、小角散乱(SAS)すなわちX線小角散乱(SAXS)または中性子散乱(SANS)を構造的生物学の主流に押し上げた。構造生物学者にとっての小角散乱の魅力は、それが広範囲にわたる多様な高分子系の まさに溶液中 で解析できることであり、それは数キロダルトンからメガダルトンまでの分子量範囲で、見かけ上無限に広がるサンプル環境で行うことができる。グローバルな構造パラメータ、たとえば回転半径 、 Rg 、最大長、 Dmax 、ならびに(体積と構造に関連する)一つの散乱体内の距離の分布は、データから迅速に得られる。さらに、小角散乱を用いて3D空間表現の低分解能像をルーチンで得ることができる。これらの構造モデリングは、事前の前提条件を入れないアブイニシオ法やX線結晶学、NMR、EM、及び相同性モデリングから得られた原子レベルのモデルの剛体モデル法(ハイブリッド法)により得られる。重要なことは、溶液環境が制御できるので、小角散乱は、サンプル条件を変えることにより生体高分子の構造応答を調べることができる。 多分散状態、例えば天然変性タンパク質および複合体または集合体形成などをSASを使用して実時間で評価することができる。これは、高解像度手法では難しい。
小角散乱の欠点の一つは、測定された散乱プロファイルが実際に目的のターゲット物質から得られていることを確実に証明することが困難であることである。すべての物質は放射線(X線および中性子)を散乱させる可能性があるため、サンプルを構成するすべての原子(高分子、水、バッファー成分、高分子、サンプル容器など)は、それぞれ測定された散乱強度に貢献する。基本的に、あらゆる小角散乱実験の成功は、十分に特徴付けされた、高品質のサンプルに依存し、サンプルはバックグラウンド散乱の寄与を正確に理解し、補正したものでないといけない。したがって、SAXSとSANSにおいてサンプルの品質を維持することは2つの技術の物理学がいかにサンプルの性質に影響するかによっている。
SAXSまたはSANSを適用して溶液中の生体高分子の構造を調べることに関心を持つラボベンチ構造生物学者にとって、SAXSとSANSを説明するかなりの量の物理学がどのようにサンプル調整に関連付けるか解釈することが難しい。 実際上必要とされることは、高品質サンプルを調整するためにどのような実用的工程が必要かを決めるのに役立ついくつかの概念を理解するだけである。 SASの物理学および数学の高度で詳細な説明は、データを分析するときや実験の設計時にますます関連性が増すが、それらは、GlatterとKratky、FeiginとSvergunと最近のSvergunらによる本がある。SASデータ収集、基本的なデータ解釈および出版ガイドラインに関する追加のプロトコルは、Skouら、Grishaev、Jacques and Trewhella、およびJacquesらの文献を参照のこと。
小角散乱の基礎:大きな意味を持つ簡単な方程式¶
SAS強度の角度依存性と、溶液中の高分子構造およびサンプルのバルク特性は非常に単純な関係で関連付ける。 サンプルがn個の独立したランダムに配向された散乱粒子を含む場合、強度は次のように表すことができる。
ここで、 \(q=4\pi\sin\theta / \lambda\) であり、θは散乱角の半分、λは入射放射線の波長である。この式は、散乱放射線の強度 \(I(q)\) が、サンプルの照射された体 積内の各個々の粒子からの散乱の合計であることを述べている。 \(I(q)\) の角度依存性はいくつかの要因に比例する。そのうち形状因子 \(P_i(q)\) は構造生物学者にとっておそらく最も興味深いものである。形状因子 \(P_i(q)\) は、実空間中の全体的な構造情報を有しており、一つの散乱体内の散乱中心間の2点間距離分布関数( \(p_i(r)\) 、図1)に関連する。
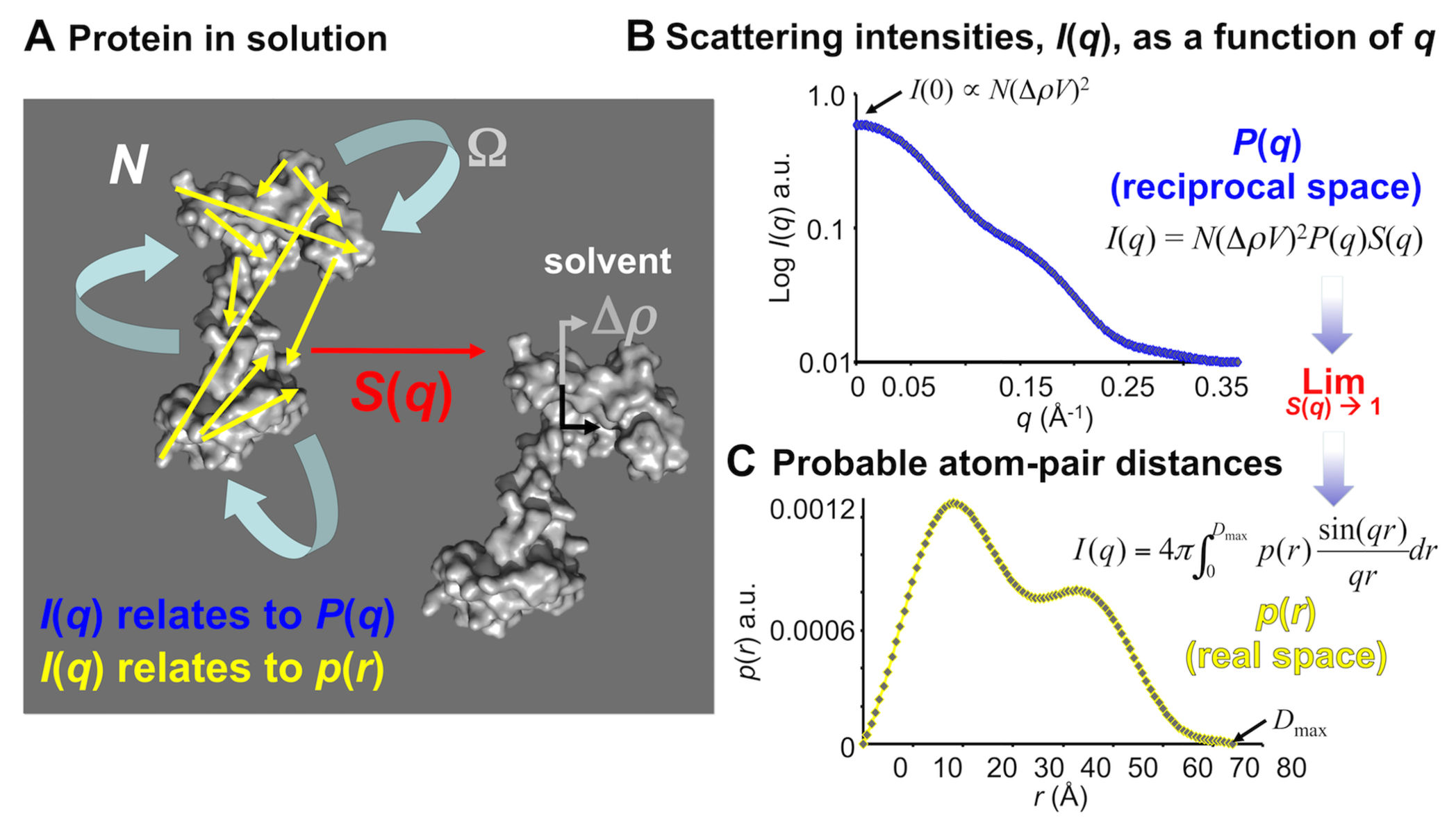
散乱の基礎 A. 溶液中の生体高分子、例えばタンパク質(灰色の塊状物として表される)は、回転運動および並進運動を受け、さらに隣接する粒子との長距離相互作用 を受ける。等方的回転摂動(Ω)する単分散サンプルから測定されたSAS強度は、散乱因子 \(P(q)\) が構造生物学者にとって最も興味深いいくつかの要因に依存する。 高分子の構造パラメータおよび低分解能モデルを得ることができるのは \(P(q)\) からである。逆空間における散乱強度の形状因子は、 実空間における2点間距離分布関数 \(p(r)\) に関係する。そして \(p(r)\) は時間平均された、(相関のある)粒子内の散乱中心間のすべてに関連する (黄色の矢印)。小角領域では、これらの相関距は溶媒中に存在しない。しかしながら、全ての原子が放射線を散乱することができるので、高分子からの \(P(q)\) を明らかにするために、溶媒散乱寄与は、サンプル散乱から正確に差分しなければならない。 散乱強度の大きさは、以下に依存する: i)試料中の粒子の数( N )。 ii)高分子体積の二乗(V2 )。 iii)散乱長密度の差、または溶媒に対するコントラスト(二乗した値(Δρ2))。 iv)隣接する粒子間の相関距離に由来する散乱(粒子間干渉、または構造因子、 \(S(q)\) )からの散乱。純度、濃度、コントラスト、 そして溶媒とサンプルとがいかに一致するかは、サンプル調製中に直接的に制御することができる。 B.SASデータは、2D検出器で収集され、半径方向に平均化されて、散乱強度の1Dプロファイル \(I(q)\) を角度θの関数として生成する。 溶媒差分後、 \(I(q)\) 対 q は、サンプル中の N (ΔρV)2および \(S(q)\) でそれぞれ重み付けされた全ての分子からの \(P(q)\) の情報を保持する。 より長距離の情報は、より低角度に現れ、その逆もまた同様である。散乱角ゼロ、 \(q=0\) では、散乱の大きさは、高分子の総体積の二乗および濃度に比例する。 もし \(S(q)\) が1に制限されている、すなわち系が無限に希薄で粒子間の影響がない場合、 \(I(q)\) 対 q の間接逆フーリエ変換で実空間 \(p(r)\) をモデリングでき、 その関数から回転半径 Rg 、最大長 Dmax 、および低分解能粒子の形状および構造を決定することができる。
しかし、 \(I(q)\) は、その他にも3つの因子に依存する。
- 各粒子の体積二乗、 \(V_i^2\)
- コントラスト(高分子とその親和溶媒との間の散乱密度の差)の二乗、 \(\Delta\rho_i^2\) である。
- 構造因子 \(S(q)\) は、粒子間の相関した動き/距離に関する情報を含む。すなわち、粒子間相互作用である。
サンプル調製に関して、サンプルの均質性、濃度およびコントラストが、 \(I(q)\) に直接的に寄与するパラメータであり、ラボベンチの工程で影響され得る。例えば、溶液中のサンプルが異なる種の混合物からなる場合、すなわち、均一に精製されない場合、混合物中の各成分は、異なる体積、コントラストおよび形状因子を有する。 結果として、式 1および図1に示すように、SASデータから抽出された構造パラメータは、混合物中の各成分の加重総和(平均ではない)を反映する。 したがって、高分子から正確な構造情報を取得し、個々のタンパク質、核酸、複合体、集合体などの3Dモデルを得るためには、サンプルは均質であり、有意な粒子間相互作用(すなわち、 \(S(q)\) を排除する)によって影響されてはいけない。 これらの条件が満たされれば、上の関係は簡単になる:
ここで N はサンプル中の均質な粒子の数密度である。 したがって、純粋なサンプルで非相互作用(希釈)条件下では、 \(I(q)\) の大きさは、粒子の濃度、体積、コントラスト、および、重要な全体の構造および形状に依存する。 ウェットラボの目的は、サンプル条件を最適化して、目的の粒子(単量体、二量体、オリゴマーまたは複合体でありうる)が可能な限り純粋であり、測定中に単分散状態で、 \(P(q)\) が散乱強度から正確に評価されうるようにすることである。 これは、濃度、コントラストおよび純度を最適化することによって達成することができる。
検討のポイント¶
生体高分子溶液散乱のサンプルを調製する際に留意すべき主な概念は、以下のとおりである。
重要
X線は電子によって散乱されるが、中性子は主に原子核によって散乱され、一般に、X線は生体高分子にはるかに大きなダメージを与える。 なぜなら、中性子よりもX線が化学変化(例えば、フリーラジカル形成)を引き起こし、結果として照射中にサンプルの状態を変化させる (例えば、架橋による凝集)可能性がある。
目的の生体高分子だけでなく、サンプル中のすべての原子(空気、水、試料、観測セル、小化学物質、緩衝成分などを含む)は、 異なる量のX線または中性子を吸収、散乱できる能力がある。検出器に到達するX線または 中性子がどこから散乱したか判別するのは不可能なので、バックグランド散乱をサンプル散乱から差し引いて、 生体高分子による散乱を明らかにする必要がある。したがって、同一条件下で少なくとも2回の測定が必要である。
i)試料(高分子+溶媒+サンプルセル)
ii)バックグラウンド(溶媒+サンプルセル)。
生体高分子が溶解している溶媒は、バックグラウンド散乱を測定するために使用される溶媒と同じである。もし、サンプル溶媒と バックグラウンド溶媒とが一致していない場合には、差分した散乱プロファイルは、生体高分子およびミスマッチした溶媒の両方を含む。
X線と中性子の両方について、生体高分子と溶媒の散乱長の差(Δρ)をコントラストという Δρがゼロに等しい場合、実質上正味の弾性散乱が得られない。 例外として、粒子内の不均一性や生体高分子の溶媒和層などに由来する弱い散乱寄与がある。SAXSでは、試料のコントラストは、生体高分子の平均電子密度および 溶媒の平均電子密度の差である。 SANSの場合、コントラストは高分子の平均中性子散乱長密度と溶媒の平均中性子散乱長密度の差である。 中性子散乱長は、生体高分子と溶媒の同位体組成に依存する。
SAXSでは、Δρを変更する唯一の実用的な方法は、サンプルの化学環境を変化させることである。 X線のコントラストは、溶媒中の小分子の濃度、 または電子密度の高い分子や重原子を試料に添加することによって測定できる(図2)。 SANSの場合、Δρは試料の同位体組成を変えることによって 変更することができる。最も豊富な水素の2つの同位体であるプロトン( \(\rm{{ }^1H}\) )と重水素( \(\rm{{ }^2H}\) )は、中性子の散乱長が大きく異なる。結果として、 Δρは、支持溶媒の \(\rm{{ }^1H_2O:{ }^2H_2O}\) 比を変更することによって、または非交換可能な水素位置で(すなわち、 \(\rm{{ }^2H}\) が官能基に共有結合し、 溶媒との迅速な交換できない)組換え高分子に \(\rm{{ }^2H}\) を導入することによって操作することができる 。
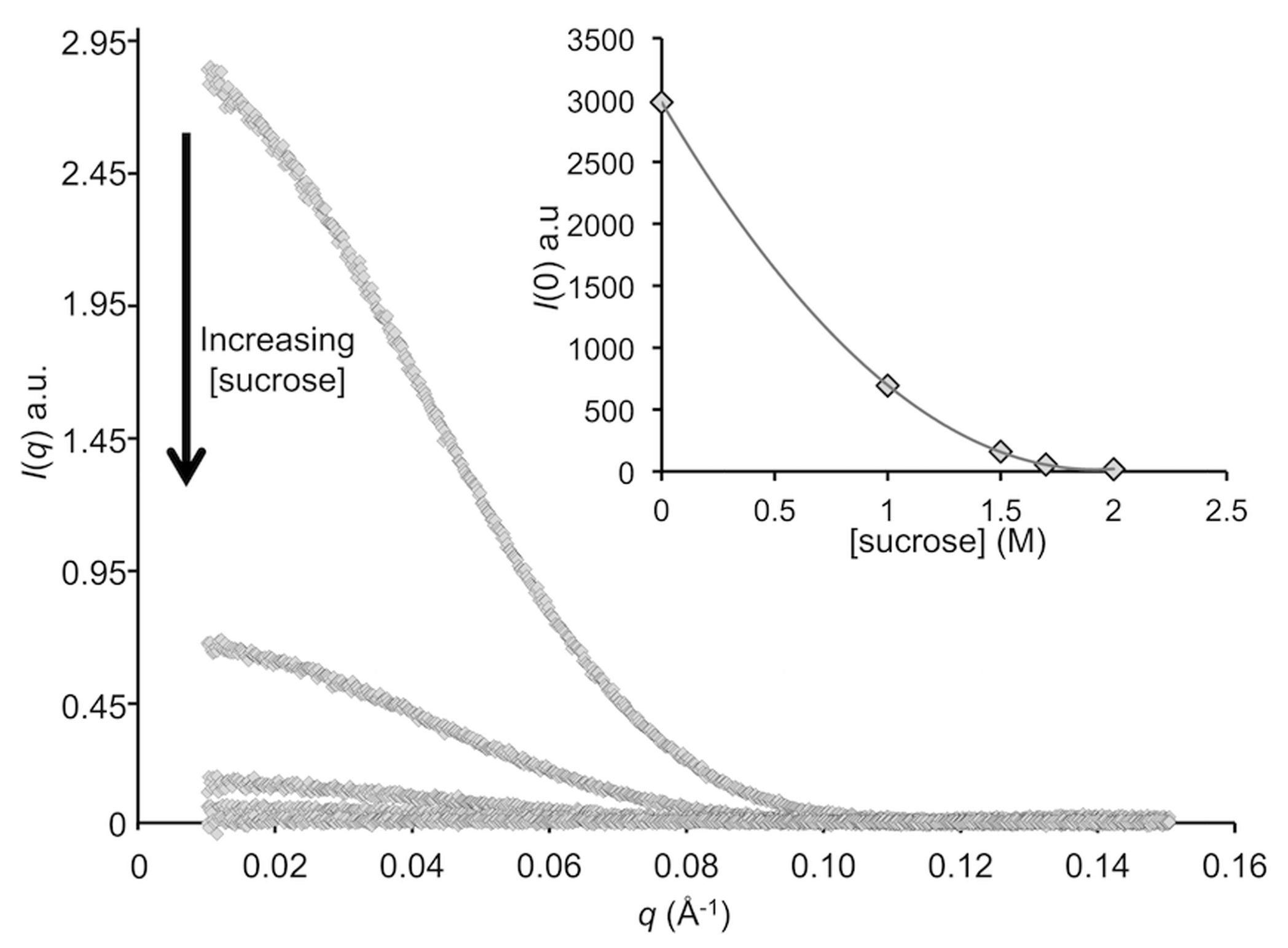
コントラストの低下(Δρ)と測定された散乱強度への影響. グルコースイソメラーゼについて溶けている溶媒中に存在する蔗糖の濃度を 漸増して収集されたSAXSデータ。 タンパク質と比較して溶媒の電子密度を増加させると、X線コントラストΔρの低下しそれにより \(I(q)\) が著しく減少する。 挿入図:ゼロ角度で計算された全前方散乱、 \(I(0)\) とスクロース濃度との間に観察される二次関係。 2Mスクロースでは、タンパク質は事実上一致しており、 すなわちΔρ= 0である。 データは、ハンブルクのDESRAのPETRAIIIのEMBL P12 BioSAXSビームライン8で収集した。
- 散乱粒子の体積が大きいほど、粒子の体積内の散乱中心間に存在する相関距離の数が多くなる。これこそが、比較的よく保存された粒子の体積内 の散乱中心間の2点間距離の相関であり、低角領域でSAS強度を生成する溶媒には存在しない。バックグラウンド差分後、散乱角ゼロでの散乱強度 \(I(0)\) は、 コントラストの二乗で重み付けされたすべての2点間相関距離からの全散乱の総和を表す。重要なことに、単分散系については、 \(I(0)\) は、 高分子の体積の二乗に比例する。
- 生体高分子の濃度を2倍にすると小角領域の散乱信号(すなわち、S/N比)が倍増する。しかし、濃度を上げ過ぎると、粒子間の最近接距離の相関が 重要となってくる。それらは、構造因子、すなわち \(S(q)\) と言われている。粒子間の親和的な相互作用は、実験データに由来する構造パラメータ、 例えば回転半径( Rg ) 、最大長( Dmax )および \(I(0)\) を系統的に増加させる。
- 反発的相互作用は構造的パラメタを系統的に減少させる。粒子間干渉は、主に低角領域のデータに著しく影響するが、 その寄与はデータの有効な領域に広がり、解釈を複雑にする。それゆえ、通常、SAS実験は低濃度で行なわれる(典型的には10mg / ml未満 すなわち、1容量パーセンテージである)。さらに重要なことは、それゆえサンプル濃度シリーズを測定し、データを無限希釈に外挿しなくてはならない。
- SAS測定は、設定された露光時間にわたって行われる。X線の場合、秒かミリ秒(synchrotron-SAXS)または数分から数時間(ラボベース光源)となる。 中性子の場合、通常は数分から数時間である。データ収集中のサンプルの安定性を確保する必要がある。
上記の概要は、SAXSとSANSの両方に適用される。 しかしながら、SANSの物理学、すなわち、中性子 - 核相互作用から生じる散乱現象は、サンプル調整に追加要件を課す。生物学的SANS実験に特有の手順に関してはより詳細に議論される。
実験材料¶
薬品¶
試薬のリストは広範であり、読者は標準的なウエットラボへのアクセスが読者にあると仮定する。例えば、細菌培養培地(例えば、Lysogeny Broth、LB52およびBox 3を参照)ならびにSDS-PAGE、タンパク質精製、サイズ排除クロマトグラフィーおよび透析などのための緩衝液を作製などが含まれる。タンパク質に関しては、目的の正しい遺伝子が適切な発現ベクター(例えば、プラスミド)にクローニングされており、大腸菌のタンパク質過剰発現株 (Box 3も参照)が得られるとする。 SANSの場合、重水は高価(1リットルあたり〜1000ユーロ)ではあるが、コントラストマッチング/バリエーション実験には重水の使用が不可欠である。
実験器具¶
一般的なウエットラボの実験装置および消耗品へのアクセスが想定されている。一連の手順の具体的な装置には、
SDS-PAGE装置(例えばBioRad製)、透析装置(例えば、SnakeSkin(商標)透析膜またはSlide-A-LyzerTMカセット)、遠心分離フィルター(微粒子を濾過するための0.1〜0.44μMの細孔サイズ、およびタンパク質濃度については公称分子量カットオフ、例えば3.5〜50kDaのもの)、高速液体クロマトグラフィー(HPLC、例えばAgilent Technologies)または高速タンパク質液体クロマトグラフィー(FPLC、例えばGE Life SciencesÄKTA)システム、サイズ排除クロマトグラフィーカラム、分光光度計(例えば、NanoDrop)または屈折計、 スタンドアローンの動的および/または静的光散乱装置、または(任意選択の)HPLCまたはFPLC-SECシステムに取り付けられたインラインサイズ排除クロマトグラフィーMALLS / RALLS(例えば、WyattまたはMalvernから)。
シンクロトロンbioSAXSビームラインまたはラボベースのSAXS機器 (例えば、Rigaku、Anton Paar、Brucker、Xenocsの)。中性子散乱の場合、SANSビームライン。このテキストに記載されている計算を実行するには、 MULCh は http://smb-research.smb.usyd.edu.au/NCVWeb/ で、 ATSAS は http://www.embl-hamburg.de/biosaxs/software.html からダウンロードできる。別途追加のオンラインツールが本文全体に記載されている。