ボックス1: インラインーサイズ排除クロマトグラフィー-SAXS(SEC-SAXS)。¶
タイムチャート:¶
緩衝液調製+カラムおよび検出器の平衡化、2〜12時間;
1×SEC-SAXS実行、30分〜2時間(SAXSビームラインとSECカラムフロー速度に依存する)。
データ処理、20分-2時間。
方法の概要。¶
インラインサイズ排除クロマトグラフィー、SAXS(SEC-SAXS)はBioCAT(Advanced Photon Source)、SWING(Soleil)、オーストラリアシンクロトロンのSAXSビームライン、ESRFでのBM29、および台湾シンクロトロンNSRRCのBL23A1を含む数々のシンクロトロンビームラインでサンプルの連続フローとして導入されている。 EMBL P12ビームライン(DESY Hamburg)では、SEC-SAXSは直角レーザ光散乱(RALLS)、UV吸収および屈折率(RI)検出器を含むトリプル検出器アレイと連動して動作し、 SECカラムの直後に設置されている。追加の検出器は、移動相流分配器を使用して並列にSAXSビームラインに組み込まれ、SAXSおよび独立して取得されたRI(UV)-RALLS測定値を直接組み合わせることができるようにしている。レーザー光およびX線散乱からの結果をRIまたはUV測定と組み合わせることにより、SECカラムから溶出する分離したサンプル分画の分子量を導出することができる。
SEC-SAXSは既に純粋な平衡系(例えば、モノマー - オリゴマー相互変換)の成分を分離、あるいは、X線露光直前のサンプルから微量の凝集物の除去するために非常に有用である。しかし、SEC-SAXSはすべてのサンプルについて「完全治癒」ではなく、また精製過程ともみなすのではなく、必要に応じてケースバイケースで適用される分析手順である。例えば、図9は、2つのサンプル1と2のSDS-PAGE結果を示しています.SEC-SAXSを使用してサンプル1の成分を分析することは不可能である。というのは、いかなるSEC-カラムの分解能をも凌駕する非常に多くのコンタミを含んでいるからである。
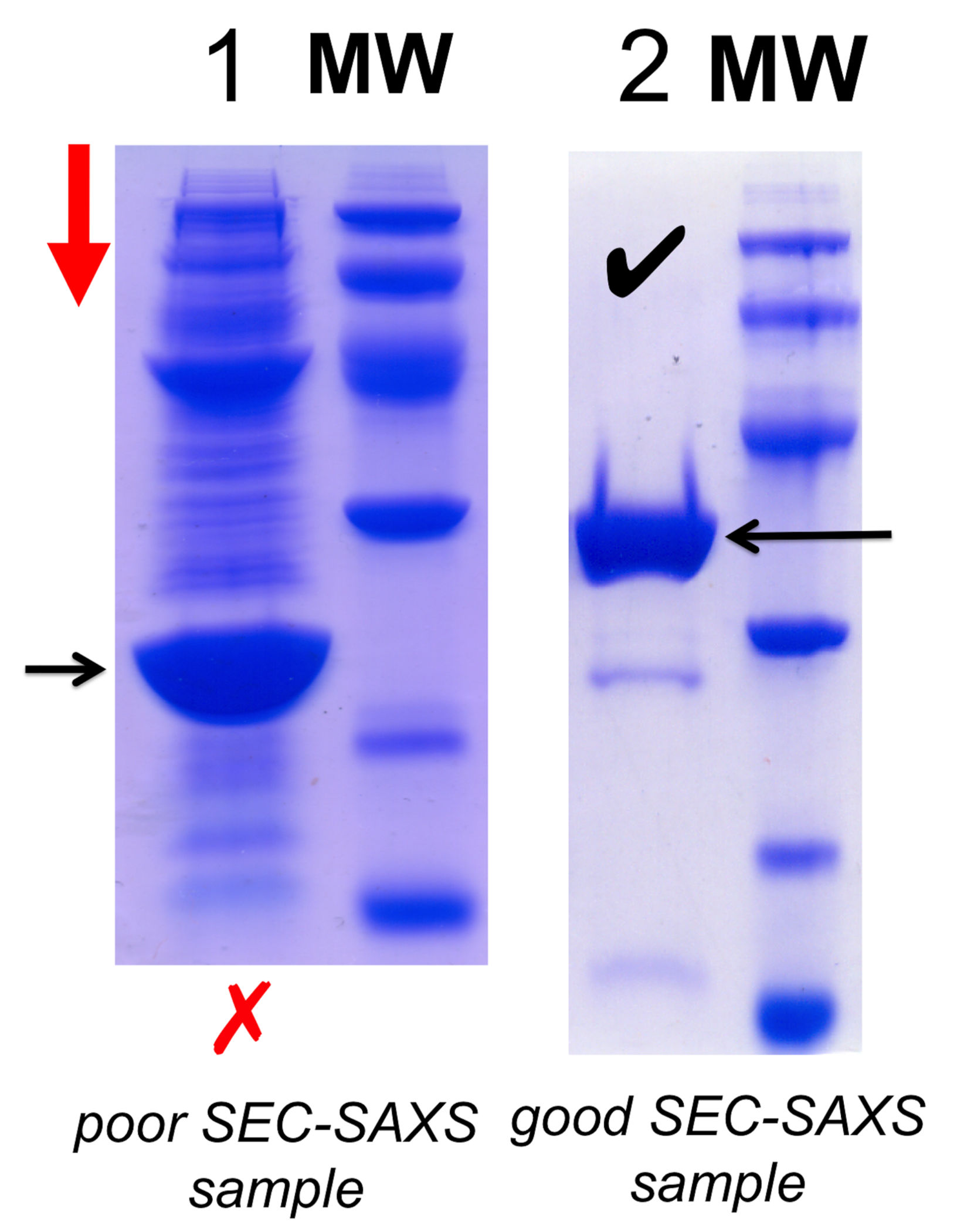
SEC-SAXSサンプルのSDS-PAGE評価。左のサンプル1は、目的の標的タンパク質(矢印)を含むが、SEC-SAXS実験を成功させるにはあまりにも多くの他の構成要素がある。 ほぼ純粋なサンプル2は、SEC-SAXSの良い候補となる。
カラム分解能は、カラムのサイズ、充填マトリックスの選択、サンプル負荷容量、サンプル流量、溶媒条件およびサンプル純度によって決まり、これら全てに対しSEC-SAXS実験(図10)の前に評価する必要がある 。
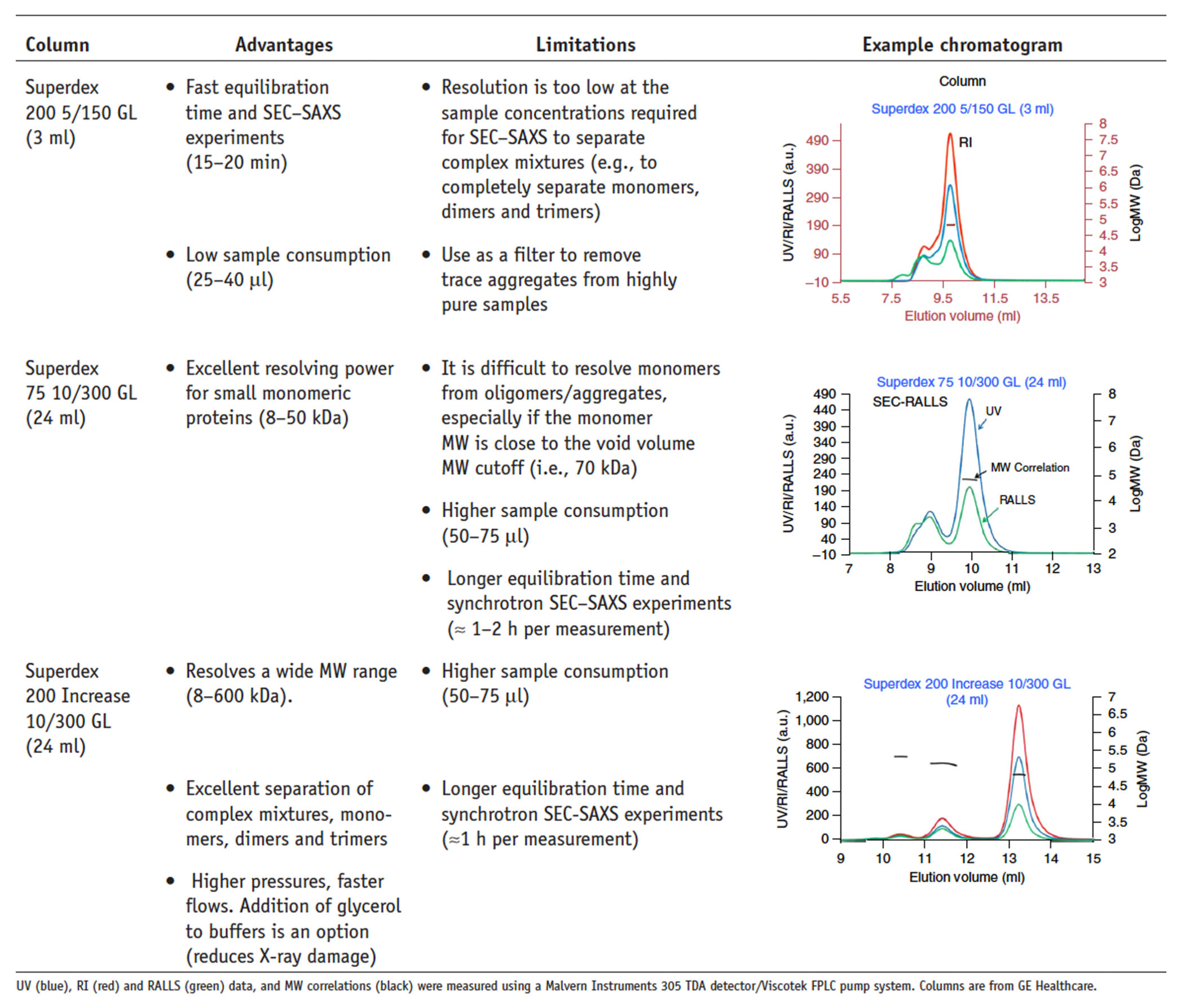
SEC-SAXSのSECカラムの分解能。 カラムの選択は、SEC-SAXSのサンプル中の構成要素を分離するために重要です。 ここでは、モノマー、ダイマーおよびより高次のオリゴマーからなるウシ血清アルブミンを、3つの異なるカラム担体を用いて分離した。 小さなS200 5/150カラムは、オリゴマーから単量体分離のレベル(最高ピーク)を示すが、これらの単量体が他の自己会合状態から完全に分離されない可能性がある。 S200 Increase は、SAXS解析のための単量体成分および二量体成分の分離を可能にするのに十分な分離されたピークを生じる。
もしカラムの分解能が損なわれた場合、すなわち溶出ピークが「互いに絡み合う」場合、SAXSデータも損なわれます。すなわち、溶出によって収集された連続したSAXSデータフレームは、連続的に変化するサンプル成分の混合物組成からの加重総和の寄与になる(式1)。
しかし、成分が十分に分離されている場合、SEC-SAXSは多分散系の構造と性質を決定するのに不可欠となりうる。予期されたSEC-SAXS実験の結果を図15に示し、 SASDBJエントリ SASDBJ3 および SASDBK3 を参照のこと。
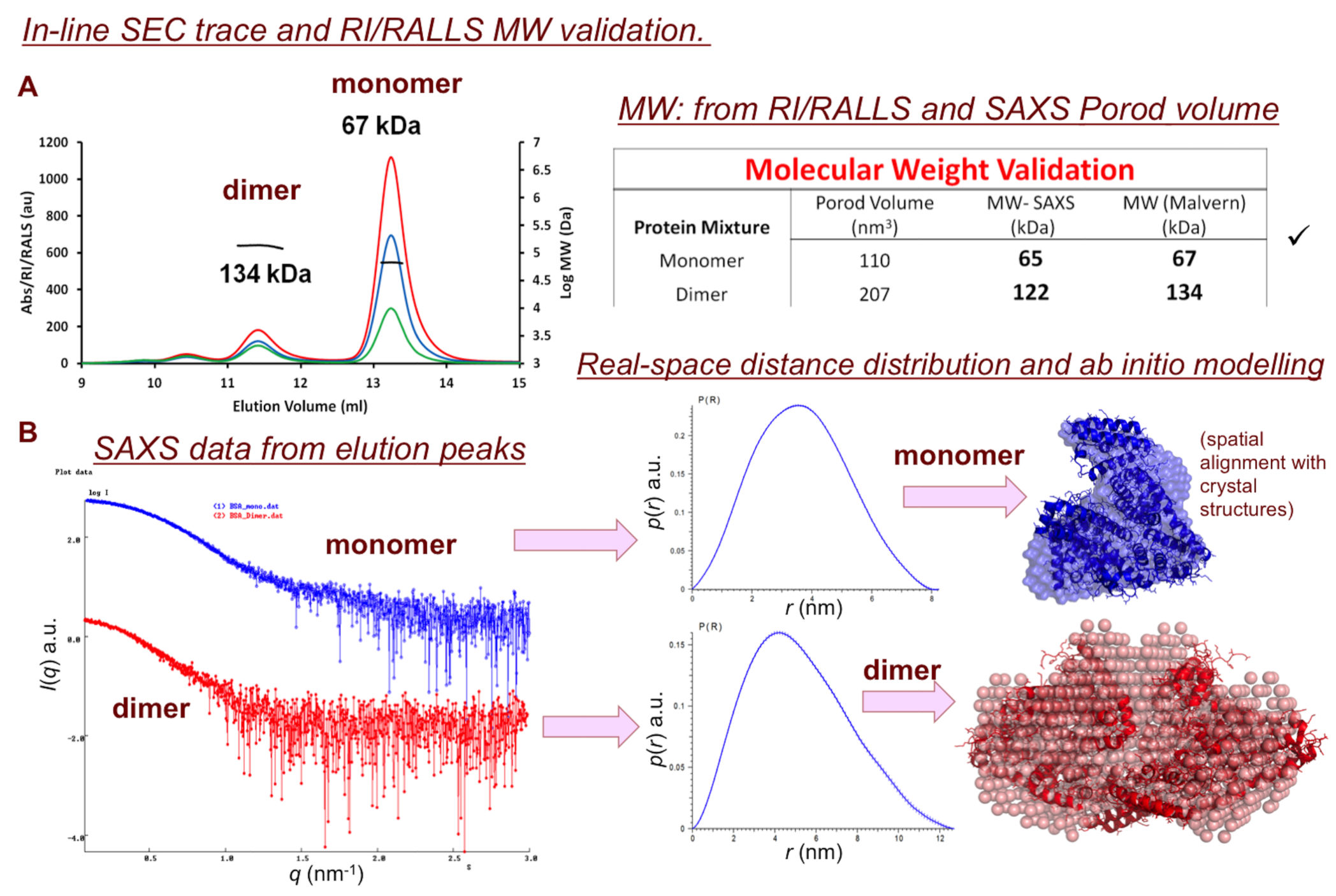
予想される結果:SEC-SAXSの成分分離。ウシ血清アルブミン単量体および二量体分離。RI/RALLS(Malvern Instruments 305 TDA検出器)で分子量確認し、サンプル中のBSA単量体および二量体画分のSAXS解析とを組み合わせたものである。 SAXS(SASBDB SASDBJ3 および SASDBK3 )の Ab initio ビーズモデルは、公開された結晶構造(Protein Databank、PDB、 3V03 )と比較される。
使用されたSEC-SAXS法は、特定のビームラインで利用可能な装置およびデータ処理ツールに依存する。 このボックスは、いかにこの実験が設定し実行されるかに関する一般的なアドバイスである。
実験材料¶
- タンパク質サンプル。 SEC-SAXSは、5-15 mg/mlで50-100 μlのタンパク質サンプルを必要とし(図9)、好ましくはできるだけ純度の高く、0.22または0.45 μmのスピンフィルターで 濾過するか、または高速で遠心分離(10分; 15 -30 000×g)して、塵または不溶性凝集物を除去する。
- SECのカラム。カラムの選択については、図10を参照のこと。
- ランニングバッファー。 SEC-SAXS実験の前後でSECカラムを平衡させるために過剰の緩衝液を調製する。ランニングバッファーは、0.45または0.22 μmの濾過および脱気が 必要です。カラムの温度が急激に変化しないようにし、平衡処理中にバッファとカラムが同じ温度になるようにすること。高輝度SAXSビームラインでは、例えば3〜5 %v/vグリセロール、1〜2mM DTTまたは1〜2 mMアスコルビン酸などの溶液添加剤をSECランニングバッファーに添加して放射線損傷を低減する必要がある。リン酸の代わりにトリスまたはHEPESを使用すると、放射線損傷を低減するのに役立つ(トラブルシューティング;図8)。
- HPLC/FPLCポンプ流量。カラムの流量を選択し、カラムをランニングバッファーと平衡化させる。 SEC-SAXSの場合、流速は通常0.25-0.35 ml/minである。流速が遅すぎると、 サンプルへのX線照射による損傷が発生することがある。ほとんどの市販のカラムでは、使用上限圧力を超えてはいけない。
- (オプション)追加の検出器。光散乱または分光光度計が利用可能な場合、分子量標準(例えば、タンパク質はウシ血清アルブミンを使用する)の 濃度(例えばRIまたはUVを使用)および光散乱強度(例えばRALLSまたはMALLSを使用)を較正する。次いで、較正された検出器を使用して、SEC-SAXSサンプル濃度を決定することができる。 UVまたはRIからの濃度値は、処理されたSAXSデータが \(I(0)\) からのMW決定のための濃度スケールに変換されることを可能にする(手順2、工程2参照)。 SEC-SAXS UV/RIをMALLS/RALLSと組み合わせると、分離されたサンプル成分の独立した MW 推定値を得ることができ、 \(I(0)\) からの MW を検証するために使用できる。
手順¶
SECカラムを、好ましくは一晩、SECランニングバッファーで平衡化する。
重要
<CRITICAL STEP>
SECカラムは、SEC-SAXS測定の前に、2〜8カラム容量のランニングバッファーを使用して、非常によく平衡化されていなければならない。 正確なバックグラウンド差分に必要な一致した溶媒に対応するSAXSデータを測定する分画を増やすには、広範なカラム平衡が必要です。
ご用心
SECカラムから流出する緩衝液(例えば、280nm)から記録された安定したUV吸収ベースラインは、カラムが実際に平衡していることを示すものではない。 例えば、150mMのNaClを含有する緩衝液は、250mMのNaClを含有する緩衝液とほぼ同一の280nmのUV吸収特性を有するが、これらの2つの溶液(異なる電子密度を有する)は、異なるSAXSプロファイルを生成する。 RIは、カラムが完全に平衡しているかどうかを評価するより敏感なツールである。
重要
<CRITICAL STEP>
SEC-SAXSのサンプルは、通常のSAXSと比較して、カラムから溶出するにつれて5〜10倍希釈されるため、かなり多く調製する必要があります。 SAXS強度における合理的な計測統計を維持し、高分子複合体の完全性を維持し、カラムの希釈効果(式2、\(I(q)\propto N\) )を克服するために高濃度のサンプルロードがしばしば必要とされる。
サンプルを適切な流速でカラムに注入してSEC-SAXS実験を開始し,同時にSAXSデータ収集を開始する。
(オプション)SAXSと並行して、UVまたはRI/UV/RALLSまたはMALLS測定を開始する。
カラム溶出液からSAXSデータを収集し、十分な数のバッファおよびサンプルフレームを測定する。 SEC-SAXS実験の開始時、終了時、および途中で溶出バッファーから SAXSデータを測定することをお勧めする。 好ましくは、SECカラムからの全溶出プロファイルにまたがるSAXSデータを収集する必要がある。
サンプルピークがカラムから溶出した後、少なくとも1つの完全なカラム容積をカラムに流したことを確認するか、すべてのサンプル成分が溶出するまで、 必ずSEC-SAXS実験が完了するまで行うこと。 SAXS実験の後、次のサンプルを実行する前に、0.1〜0.25カラム容量の追加のランニングバッファーをカラムに流す。 この追加の洗浄は、先行するサンプルからの小分子の全てがカラムから吐き出され、次のSEC-SAXS実験のバックグラウンド散乱を汚染しないことを保証する。
各SEC-SAXS実験後にSAXSサンプルセル(例えば、サンプルキャピラリー)が清浄であるかどうかを評価する。カラム溶出の始めと終わり(例えば、相関マップを使用して) で緩衝液から測定された(差分しない)SAXSプロファイルを比較する。違いがある場合は、水ー洗浄溶液ー水のサイクルを使用してSAXSサンプルセルを洗浄すること。
洗浄溶液の3つの例は、
- 6M塩酸グアニジン、pH6.5
- 20%v/v酢酸
- 10%v / vエタノールを含む2%v/vのHellmanexIII。
SEC-SAXS実験のバックグラウンド散乱に対応するSAXSデータフレームを選択する。 これらのフレームは、SEC-カラムを通過した溶媒/バッファーから測定した散乱強度から 選択することができる。 これらのフレームは、必ずしもそうとは限らないが、サンプルの溶出ピークに近いものであろう。
重要
<重要なステップ>
最初のデータフレームに対するサンプル溶出後のSEC-SAXSバッファーの散乱強度の系統的な増加は、サンプルキャピラリーの汚れが発生したことを示し得る。 キャピラリーの付着は、しばしば、X線ビームを通って流れるサンプルの放射線損傷の影響を受けやすい成分がキャピラリー表面に凝集して結合するによって引き起こされる。 連続するSEC-SAXS実験間にサンプルキャピラリーを洗浄して、内部キャピラリー壁上の凝集した物質の蓄積を低減することが推奨される。 キャピラリ上の集積は、正確なバックグラウンド除去を不可能にし、その後のSEC-SAXS試行のすべてを汚染します。
各SEC-SAXSデータフレームからバッファ散乱を差し引く。差分したデータの Rg および \(I(0)\) を、例えば、AUTORG を使用して計算することによって、 サンプル溶出ピークに対応するフレームを同定する。いかなる平均化の前にも、溶出ピーク近傍から取得された各フレームデータが過大差分または過小差分になっていないこと(図7)、(濃度などで)スケールした後各フレームが統計的に類似していることを確認する。
<オプション>カラム溶出をモニターするために追加のUVまたはRI検出器を使用した場合は、検出器からの濃度 c mg/mlをSAXSの \(I(0)\) に相関させ、 溶出成分の MW を計算する。(UV)RI-RALLSまたはMALLS検出器を使用する場合は、光散乱から MW を計算し、SAXS \(I(0)\) から得られた MW を確認する。これらの検出器が利用できない場合は、SAXSデータから計算された散乱体積から MW (タンパク質サンプルの場合)を推定する(手順2、手順2を参照)。
ご用心
均一で単分散で非干渉性の粒子の場合、\(I(0)/c\) 、 MW と Rg は一定であろう。しかし、SECに及ぶSAXSデータから \(I(0)/c\) 、 MW および Rg の一定値を得ることは、成分が均一で単分散であることを必ずしも意味しない。これらの結果は、初期サンプルおよびカラム分解能の純度に依存する(図9および図10)。例えば、ウシ血清アルブミンは、SECの前に溶液中で混合物として存在し、(通常のSAXS測定を使用して)一定の \(I(0)/c\) 、 MW および Rg 値を与える。もし、この混合物が、劣化したあるいは誤って選択されたSECカラムで十分に分離されない場合も、SEC-SAXSデータが溶出ピークで一定の \(I(0)/c\) 、 MW および Rg を与えると考えられる。したがって、SEC-SAXSの前に、選択されたカラムのサンプル成分の分離能を試験するためにサンプルに対してSECを実施し、必要に応じて分離を最適化するために溶媒条件(例えばpH、塩濃度)を変更することが推奨される。