手順2:サンプル濃度と分子量の定量的ガイド¶
概要:SAXSにおける定量ガイド¶
SAXSのサンプルを調製する際の多くの考慮事項の中の1つに、実験に必要なサンプルの量がある。経験則として、 標準の放射光SAXSのサンプル濃度は、通常0.1〜5.0mg.ml-1のオーダーである。放射光SEC-SAXSについては、 SECカラムによる希釈効果を補うために5〜15mg ml-1(またはそれ以上)が必要となり得る(ボックス1参照) 。 これは、多成分複合体の会合状態を維持する際に重要となる可能性がある。光量がはるかに劣る実験室用X線源の場合、 標準測定のための最低濃度は通常3〜10mg / mlである。実験室ベースの実験のサンプル濃度を濃くするのは データのS/N比を改善するために必要で、特に散乱角が大きくなるにつれ粒子散乱強度が劇的に減少する。 もし、サンプル濃度およびX線フラックスが低すぎる場合、散乱プロファイルの広角領域は、 バッファ差分後ノイズに埋もれてしまう。これらの領域は中分解能の原子対距離に関する情報 (例えば、2nm以上の分解能でのドメイン間の配置など)を含む。散乱サンプルの濃度の決定は、STEP 1を参照のこと。
1回のSAXS測定には、通常、5〜30μlのサンプル(SEC-SAXSの場合は50〜100μL、ボックス1)が必要であるが、 マイクロ流路技術が導入されればこれらの量は減少する可能性がある。しかし、どのぐらいの体積で測定できるかと、 どのくらいの体積が再現性のある実験に必要なのかは全く異なる可能性がある。例えば、実験に先立ってサンプルを 溶媒と一致させる必要があるということは、小さな容積での作業が現実的でないことを意味する。 100〜200μlの サンプルを調製することは、それを自信を持って取り扱うのに十分な材料を提供し、例えば一連の希釈、すなわち 濃度希釈シリーズをセットアップするのに十分なサンプルを提供する。
- 散乱強度(式2)に対する \(S(q)\) の影響を評価し、多成分複合体の会合状態(完全に複合体が形成されているかどうか)を評価するために、 一致溶媒を希釈剤として使用してサンプルの濃度希釈を行うことが常に推奨される。
これらのサンプル量の手引きには常に例外がある。例えば、 \(S(q)\) が重要でない限り、高分子または複合体によっては、 非常に高いサンプル濃度(10〜20mg / ml以上)で分析することが可能であり得る。高分子量(例えば、300kDa以上) の巨大分子、または長時間の露光中安定している高分子なら、低濃度(1mg ml-1 )でも実験室SAXS源から 高品質データを得ることが可能である 。
ステップ1:正確なサンプル濃度決定¶
サンプル処理手順を監視するための有用な指標であるとは別に、正確なサンプル濃度測定は、SASデータの評価にとって重要だ。 SASデータから高分子の分子量を得ることは、最も重要な品質保証ステップの1つを担っており、一つの散乱プロファイルとあるサンプルをつなぐ。 そして、サンプル濃度は±10%誤差以内に正確に決定する必要がある。 ポリヌクレオチドについてはオプションAを、 タンパク質についてはオプションBを用いてSASサンプル濃度を評価する。
オプションA:核酸濃度の評価。¶
核酸は260nmでUV光を非常に強く吸収するので、一連の核酸サンプルの濃度希釈シリーズを分光光度計の比例応答領域の中で調整する。 例えば、1mg/mlでの二本鎖DNAは、1cmの光路長セルを使用してA260が約20であるので、例えば、10,20,40および80倍の希釈を行う。 NanoDrop分光光度計(Thermo Scientific)は、より短い経路長を有し(0.05〜1mm)、より高濃度のポリヌクレオチドを希釈することなしに 記録することができる(約15mg / mlまで)。
核酸サンプルのA260nmでのUV吸光度は適合する溶媒で分光光度計をゼロにしてから測定する。
A260 nm吸光度の測定値を経路長(cm単位)か吸収係数のいずれかで割ること:
. 二本鎖DNA(約0.020(μg/ ml)-1cm-1)、
. 一本鎖DNA(約0.027(μg/ ml)-1cm -1)または
. 一本鎖RNA(約0.025(μg/ ml) -1cm -1)
そして、希釈倍率を乗じて核酸濃度の近似値を得る
オプションB:タンパク質濃度の評価。¶
タンパク質の場合、アミノ酸分析(AAA)がおそらくタンパク質濃度を決定する最も正確な方法であり、タンパク質濃度分析を較正するためのデータを提供するが、完了するまで数日から一週間かかることがあり、熟練した人員や特殊施設にアクセスする必要がある。したがって、我々はタンパク質の濃度を280nmでの吸光度を使用して分光学的に測定するか、あるいは屈折率を使用して測定することをお勧めする。屈折率測定は、若干の変更により、核酸の分析にも使用することができる。結合アッセイ色素(例えばブラッドフォード試薬)の使用は、色素アッセイが(別の技術と比較して)自信を持って標準化されている場合を除いて、タンパク質濃度を決定するのには一般にあまり正確ではない。較正用の既知量の粉末状タンパク質の濃度を推定することは、粉末タンパク質試料にしばしば付随する未知の量の塩および他分子の存在により、一般に困難である。
マッチした溶媒を用いて分光光度計をゼロにした後に、タンパク質の280nmでの吸光度を測定してタンパク質濃度を評価する。 吸光度の読みを光路長とタンパク質吸収係数で割る。吸光係数は、タンパク質のアミノ酸配列から計算される(例えば、 Protparam を使用する:http://web.expasy.org/protparam /)。
重要
<重要なステップ>
分光光度法を用いて生体高分子濃度を評価する場合、支持溶媒中の時間依存性または化学変化を考慮する必要がある。 ジチオスレイトール(DTT)のようなチオール還元剤は、それらが酸化を受けるときにそのUV吸収特性を変化させうる、あるいは染料に基づく分析に直接干渉しうる。 DTTおよび代替のチオール還元剤、β-メルカプトエタノールの両方は、pH7.5より高いpHで比較的短い半減期を有し(温度に依存して約1-20時間)、そして、DTTは、Zn 2+ のような生物学的に関連するいくつかの金属イオンに対してキレート剤として作用し、その結果吸収特性を変化させ 濃度推定値を混乱させる。 TCEP-HCLは、はるかに安定した優れた代替物であり、A280 nmの読み取り値には無視できる影響しかないが、中性pHのリン酸緩衝液中で有効性が損なわれる。 非常に酸性であるので、TCEP-HCL添加後に溶媒のpHを意図した値に戻すように注意する必要がある。
注釈
- TCEP-HCL
Tris(2-carboxyethyl)phosphine Hydrochloride。ジスルフィド結合を切断する還元剤。特長としては、毒物非該当、還元力が強い、無臭、広範囲のpHで使用可能などである。
代換えとして、タンパク質サンプルについて屈折率(RI)測定を実施する。 屈折率はタンパク質濃度を評価するには極めて有用なツールである。というのは、タンパク質の 屈折率増分(約0.185ml/g)はA280nm吸光係数とは異なり、アミノ酸配列組成の変化に対して比較的安定なであるからである。 RI増分は、核酸(DNA:約0.17ml/g、RNA:0.17-0.19ml/g)についても応用できる。 それゆえ、RIは、例えば、A280nmの吸光係数が小さいタンパク質またはタンパク質/ DNA複合体など、濃度を決定するためにより有用であろう。
ステップ2:分子量( MW )分析¶
SASデータから溶液中の生体分子の分子量を決定する方法をこの手順書に含めておこう。 散乱粒子の分子量は、試料濃度とGunier解析により外挿された散乱角ゼロにおける前方散乱強度 \(I(0)\) および計算されたもっともらしい実空間2点間距離分布関数 \(p(r)\) 対 r を組み合わせて評価できる。 オプションA(SAXS)およびオプションB(SAXSおよびSANS)に記載されている手順は、 両方とも濃度に依存する方法である。代換えとして、SAXS、特にタンパク質の場合 濃度-非依存性分子量推定は、散乱データから計算することができる散乱体積 V から得ることができる。非常に非等方的なタンパク質、揺らいだタンパク質の分子量評価の際には補正が必要であろう。 V からの MW および \(I(0)\) からの濃度に依存した MW の両方を比較することはしばしば有用である。 V からの MW にはいくつかの方法が利用可能である。その中には、Fischer et al.(SAXS-MoW http://www.if.sc.usp.br/~saxs/)、RamboとTainerの「相関体積」および体積ベースの分子量決定を用いた ATSASソフトウェアスイートの DATPOROD (ここで、タンパク質の場合、\(V_{DATPOROD}/1.6\sim MW\))。SAXSデータにフィットする(例えば、 DAMMIF を使用)タンパク質のab initioダミー原子モデル( DAM )から得られた体積も、タンパク質 MW を推定するために使用することができる。一般的な経験則で \(MW_{protein}=V_{DAM}/2\)。光散乱測定(SLS / DLSまたは組み合わせSEC-MALLSまたはSEC-RALLS)からの独立した MW 結果を取り込めば、 SAS測定から得られる間違いなく最も重要な全体パラメータの1つである MW をさらに検証するのに役立つ(ボックス1参照)。
注釈
当研究室では、
- \(V_{DATPOROD}\)
- Porod体積
- \(V_{DAM}\)
- Beads体積
と読んでいる。
オプションA: I(0) からの MW 、SAXS。¶
SAXSについては、サンプルをスケーリングして濃度依存 I(0)_MW 分析を行う。これは濃度がわかっている標準タンパク質の散乱データにサンプルの散乱データをスケーリングすることにより行われる。 この標準タンパク質は、試料と同様のコントラストを有するべきである(例えば、水溶液中のタンパク質試料についてはリゾチーム標準を使用する)。
MW 標準と対応する溶媒ブランクからSAXSデータを収集する。温度、露光時間、サンプル(キャピラリー)セルなど同じ実験セットアップを使用すること。 散乱データを一次元化して(例えば、2Dデータを半径方向に平均して1Dデータにする)、標準と溶媒の差分していない \(I(q)\) 対 q プロファイルを作成する。
注釈
点光源ではない装置、例えばKratkyカメラでは、関連するビーム形状補正をSAXSデータに適用する。
標準タンパク質の散乱から溶媒の散乱を差し引いて、溶液中の標準高分子の \(I(q)\) 対 q 、1D差分プロファイルを得る。
未知 MW およびそれに対応する一致した溶媒がサンプルについてデータ収集手順を繰り返す。その溶媒を用いて試料を希釈して濃度希釈シリーズ(例えば、 8,6,4および2 mg/ml)を作る。 データを処理して、溶液中のサンプルタンパク質の \(I(q)\) 対 q 、1D差分プロファイルを得る。 必要に応じてビーム形状補正を適用する。
Guinier近似(例えば、 AUTORG を使用)または計算された実空間2点間距離分布関数 \(p(r)\) 対 r の面積(例えば、 AUTOGNOM を使用) を使用して、標準高分子および試料高分子の \(I(0)\) を計算する。
以下の式を使用して、標準に対するサンプルのMWを決定する
\[MW_{sample}=\dfrac{I(0)_{sample}}{I(0)_{standard}}\times \dfrac{c_{standard}\Delta\rho^2_{standard}\nu^2_{standard}}{c_{sample}\Delta\rho^2_{sample}\nu^2_{sample}} \times MW_{standard}\]正確に記録された濃度は c (w/v 単位、mg/ml)であり \(\nu\) は部分偏比容(cm3 /g)である。
重要
<重要なステップ>
もし、 \(\Delta\rho_{standard}=\Delta\rho_{sample}\) であり、標準とサンプルの \(\nu\) が類似している場合(タンパク質SAXSデータをある標準タンパク質のものに対して標準化する場合はほとんどがそうである)、上式の \(\dfrac{\Delta\rho^2_{standard}\nu^2_{standard}}{\Delta\rho^2_{sample}\nu^2_{sample}}\) の比は〜1であるため、コントラストまたは部分偏比容を決定する必要はない。しかしながら、標準タンパク質がサンプルと比較して異なるコントラストまたは部分偏比容を有する場合、例えば標準タンパク質とDNAサンプルとを比較する場合、またはグリセロール中のタンパク質サンプルと比較する場合、その比はもはや1ではない。これらの状況下では、 \(\Delta\rho_{standard}\) および \(\Delta\rho_{sample}\) の両方を計算することが必要であろう。その計算では、対応する溶媒中におけるサンプルに対する標準サンプルの電子密度差ならびにすべての \(\nu\) の差を考慮に入れなくてはならない。コントラスト計算は、 MULCh の Contrast モジュールを使用して実行できる(ボックス2)、あるいはもし原子構造が利用可能であれば、 CRYSOL を使用する。 Contrast を用いてを部分偏比容を推定することもできるし、 NucProt を使ってタンパク質やRNAを計算することもできる。http://geometry.molmovdb.org/nucprot/
サンプル濃度を変えた時に、サンプルの MW が系統的に減少または増加することが観察されるかどうかを評価する。 濃度上昇でのサンプルの MW の増加は、濃度依存性の オリゴマー化または凝集の徴候である。 濃縮上昇が典型的な排除粒子間干渉によって引き起こされる場合、サンプルの見掛け分子量は有意に減少する。
\(I(0)\) から実験的に決められたサンプルの MW と( Protparam を使用して)アミノ酸配列から計算される予想される MW を比較する。 結果を使用して、サンプルのオリゴマー化または凝集状態を評価する。
<オプション>独立した光散乱実験(例えば、SLSまたはSEC-MALLS / RI)、またはタンパク質については、SAXSデータから計算された散乱体積に基づく濃度非依存MWを測定値とクロスチェックする。
重要
<重要なステップ>
SAXSデータの MW 較正のために選択された標準タンパク質は、X線ビーム中で安定していなければならない。すなわち、放射線損傷の影響を受けてはならない(トラブルシューティング参照)。 標準のX線誘導凝集は標準 \(I(0)\) を増加させ、試料の MW の過小評価をもたらす。 さらに、標準サンプルは反発的な相互作用に過度に影響されてはいけない。その場合、 標準 \(I(0)\) の大きさが減少し、サンプルの MW を過大評価する。 もし粒子間相互作用がわからない場合は、濃度系列からSAXS測定を行い、 \(I(0)/c\) vs c をプロットする。 \(S(q)\) が無視できる場合、\(I(0)/c\) の値は(誤差内で)比較的一定である。 有意な正の勾配は正の \(S(q)\) を示す。例えば、最悪の場合凝集に当たる。 負の傾きは、負の \(S(q)\) 、すなわち反発相互作用を示唆する。
オプションB:SAXSおよびSANSの \(I_{water}(0)\) からの MW 。¶
サンプルの散乱データ \(I(q)\) を単位cm -1 を有する絶対強度に変換し、SAXSの \(I(0)\) MW 分析を行う。 SAXSの場合、絶対強度測定は、 通常、水からの散乱を基準として使用して行われる。
注釈
ちなみに、絶対強度={散乱光強度}/{入射光強度} のことである。
キャピラリー(またはサンプルセル)内の純水からSAXSデータを測定し、差分をかけていない1D散乱プロファイルを取得する。
純水の測定に使用されたものと同一で空のキャピラリー/サンプルセルから \(I(q)\) 対 q を測定する。空のキャピラリー/サンプルセルが完全に乾燥していること、 およびX線の露光時間と温度が純水の測定と同じであることを確認する。
純水の散乱から空のキャピラリー/サンプルセル散乱の寄与を除き、純水のみの差分 \(I(q)\) 対 q のプロファイルを得る。
注釈
差分する前に透過率を補正する必要がある。ただし、BS位置で強度をモニターしているのなら透過率込みの量が出力されているのでそれでそのまま規格化できる。
水の前方散乱強度の実験値、実験値 \(I_{water}(0)\) 、すなわち「装置値」を記録する。水の前方散乱を計算する簡単な方法は、中〜高角 q の領域、 すなわち水のSAXSプロファイルが「フラットな散乱領域」における \(I(q)\) の平均の大きさを決めることである (補足情報の表計算シート:
MW_from_absolute_scale.xlsxを参照のこと)。注釈
純水の広角領域の散乱断面積 \(\Sigma\) は等温圧縮率 \(\chi_T\) と密度 \(\rho\) に依存し、以下の式で表される。
\[\dfrac{d\Sigma}{d\Omega}=I_{water}(0)_{standard}=\rho^2kT\chi_T\]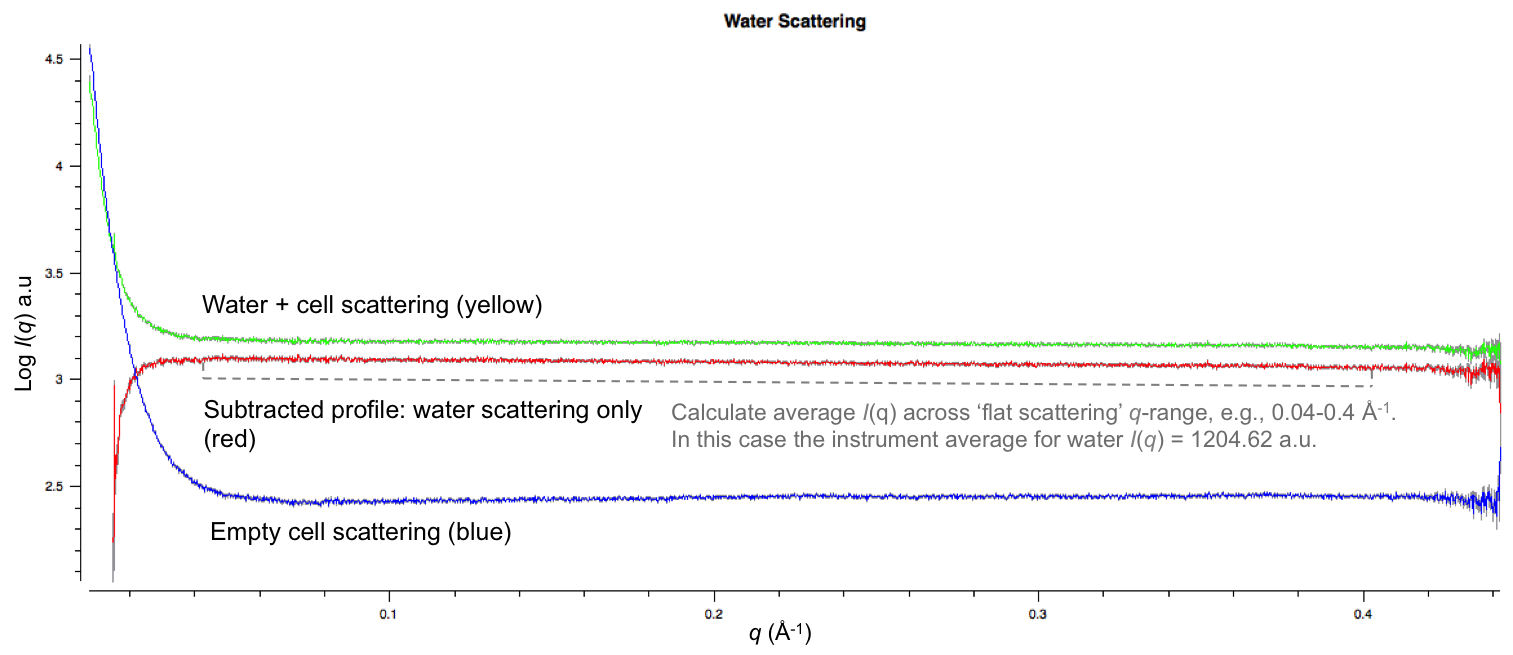
次に、純水測定と同じ温度、キャピラリー/サンプルセルでサンプルと一致する溶媒ブランクからSAXSデータを収集する。
サンプル+セルから溶媒+セルを差し引いて、サンプルの差分 \(I(q)\) 対 q プロファイルを得る。サンプル高分子の散乱強度 \(I(q)\) に次の比を掛けることによって絶対的なスケール(cm-1 )に散乱を変換する:
\[\dfrac{I_{water}(0)_{standard}}{I_{water}(0)_{experimental}}\]ここで、 \(I_{water}(0)_{standard}\) は、特定の温度での水からの既知の前方X線散乱 \(I(0)\) である (
MW_from_absolute_scale.xlsxのシート2を参照)。298Kでは \(I_{water}(0)_{standard,298K}=0.01633 \,\rm{cm^{-1}}\) である。 サンプル中の高分子の \(I(0)\) を、絶対スケールのSAXSデータからGuinier近似または \(p(r)\) vs r など標準的な方法を使用して計算する。もし \(I(0)\) をcm-1 の絶対スケールに変換し、 c をgcm-3 で求めると、高分子の分子量は次の式で評価することができる。
\[MW_{sample}=\dfrac{I(0)_{sample}N_A}{c_{sample}(\Delta\rho\nu_{sample})^2}\]ここで \(N_A\) はアボガドロの数であり、 \(\Delta\rho\nu_{sample}\) は高分子のコントラスト( \(\Delta\rho\) 、cm-2 )と 部分偏比容( \(\nu_{sample}\) 、cm3 g-1 )の積である。補足情報スプレッドシート
MW_from_absolute_scaleを参照してください。 さらに詳しい説明はBox 2を参照してください。サンプルの入射光束に対する散乱強度を正規化することにより、透過率と機器の形状補正して、SANSデータの絶対スケーリングを実行する。 異なるSANS装置は、 異なる手順で機器の絶対較正を行う。 幸いなことに、ほとんどの施設でSANSデータをcm-1 で提供しているため、追加のデータスケーリングの必要はない。 式4でコヒーレントな中性子散乱コントラストで \(\Delta\rho\) を置換して、式と同じ関係を用いて \(I(0)\) から MW を決定する。 (Contrast によって計算される、ボックス2を参照)。
注釈
手順2のタイムチャート。
核酸および/またはタンパク質の濃度測定のために、サンプルあたり1〜2分(分光光度計でブランク測定することを含む)。 SAXSを用いた MW 推定値については、 MW 標準(使用される場合)を作る時間を設定する。例えば、リゾチームは、一晩透析/緩衝液交換が必要な場合がある。通常のSAXS測定に必要な時間は、2〜10分(高輝度シンクロトロンSAXS)から1〜 4時間(実験室ベースのX線源) までで、サンプルと溶媒のロード、サンプルと溶媒のデータの収集、測定間のサンプルセル/キャピラリの洗浄と乾燥を含む。コントラストを計算するのには MULCh を用いて約5分かかる(ボックス2)。