手順1:SASサンプルの純度、品質、および溶媒ブランクの準備¶
概要¶
散乱データの健全な解釈のためには、分析中の物質が純粋であり、有意なレベルのコンタミがないことが重要である。散乱プロファイルは、溶液中の各粒子からの散乱の和(式1)を表すので、
コンタミの存在が試料の散乱強度に加わる。これらのコンタミは、汚染物質の濃度および体積二乗に比例する大きさで \(I(q)\) に寄与する。例えば、2%の100kDaタンパク質(15kDaタンパク質またはそれ自体と相互作用しない)を含む98%に精製された15kDaタンパク質からなるサンプルは、100%純度の15kDaサンプルから予想されるもののほぼ2倍の前方散乱強度を生じる(図3)。
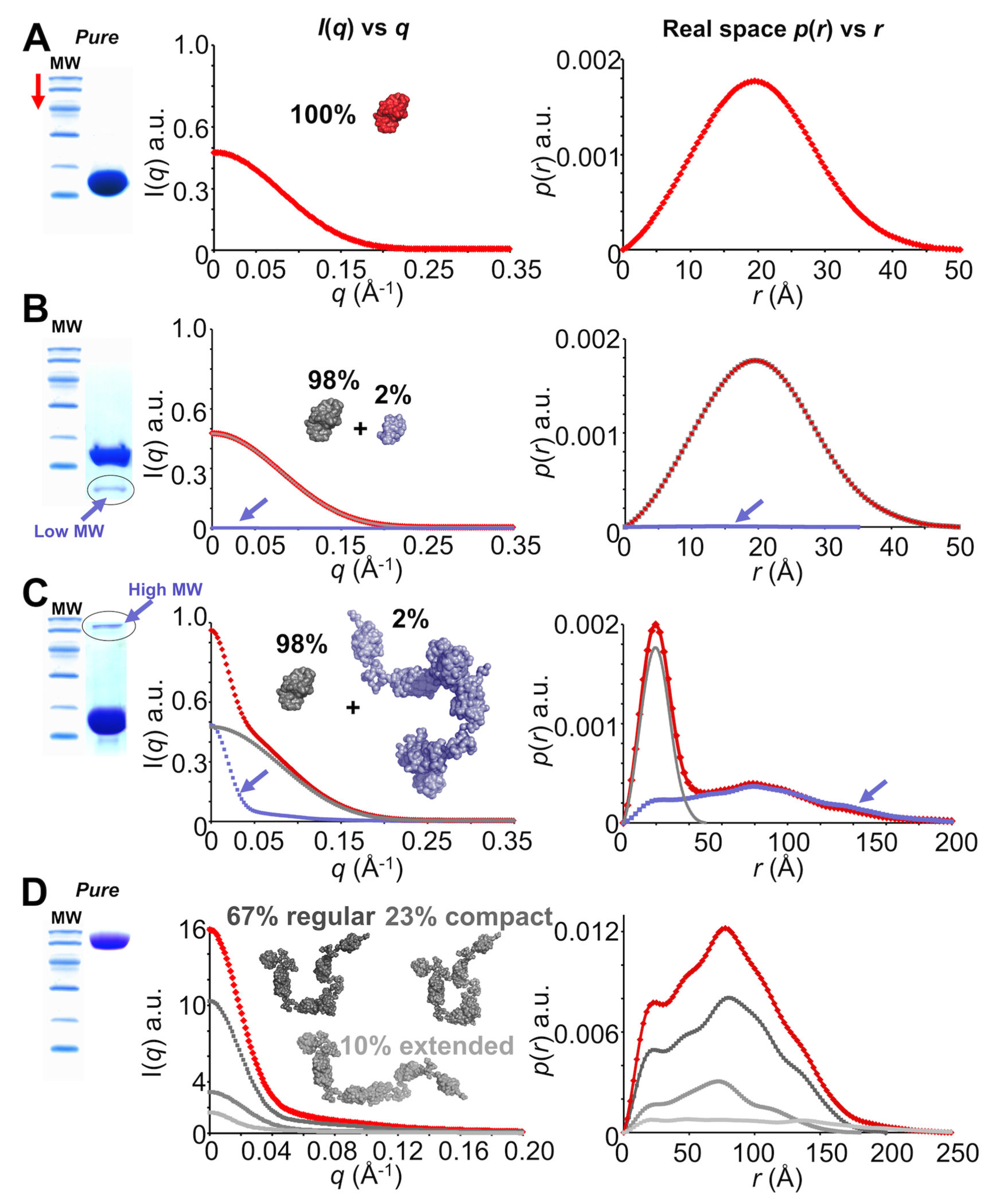
サンプル純度およびコンタミ(SAXSデータおよびSDS-PAGEはシミュレーション) A.単一のサンプルからの理想的な結果。純粋な単分散14kDaタンパク質集団内の各個体からの散乱は、加算されて全散乱曲線(赤色)を生成する。 その曲線からが単一粒子内の実空間二点間距離分布を表す \(p(r)\) 対 r をモデリングすることができる B.あまり理想的ではない状況。コンタミが存在する場合、全散乱(赤色)は、それぞれの異なる成分からの散乱の、その体積の二乗および濃度に 比例した総和となる。ここでは、低分子量(MW)のコンタミ(約5kDa、サンプルの2%、青色)が14kDaタンパク質サンプル(灰色)に存在する。 しかし、低分子量コンタミによる散乱の全体に対する寄与は小さく、 \(I(q)\) 対 q または \(p(r)\) 対 r にはあまり影響しない。 C.避けるべきこと。高分子量コンタミは \(I(q)\) 対 q (赤)に悲惨な結果をもたらす。微量の〜100kDaタンパク質による散乱寄与(青色)は、 標的14kDaタンパク質が純度98%(灰色)であっても \(I(0)\) を2倍にする。 二点間距離分布関数 \(p(r)\) 対 r に対する効果は、 それが14kDaタンパク質と100kDa汚染物質との加重和であるので顕著である。 D.特殊なケース:柔軟性。 100kDaのタンパク質は、純粋で単量体である。しかし、タンパク質は柔軟性があり、3つの主な集団から構成される場合、 散乱から決定された総 \(P(q)\) (赤色)は各集団からの \(P(q)\) (灰色の陰影)の総和となる。例えば、伸びた状態が全母集団のわずか10%を構成していても、 測定された二点間距離分布関数 \(p(r)\) 対 r (赤)の最大長 \(D_{max}\) は、最も伸びたされた状態(薄灰色)の \(D_{max}\) に等しい。 注:この図に使用されているSDS-PAGEゲルおよび散乱プロファイルは、説明のためのものであり、実際のデータを表すものではない。
したがって、凝集体およびより大きな粒子に集合する小さな汚染粒子の凝集体を含む高分子量成分の存在、動的非平衡系のオリゴマー化、あるいは 放射線誘発または時間誘発凝集などはデータの解釈とモデリングを非常に複雑にする。 SASの実験を成功させるには(特に、ビーズモデリング または剛体モデリングによるデータにフィットする3D空間モデルを作成する場合、試料中の生体高分子は可能な限り純粋であり、単分散であり、 粒子間干渉効果がないことが不可欠である。手順1では、まずSAS実験の前にサンプル品質を評価する方法(手順1)、 次に測定のためにサンプルとバッファを準備する方法(手順2)について説明する。
ステップ1:SASの実験に先立つサンプルの純度と品質の評価¶
生体高分子SASの技術は、高品質のサンプル調製および特徴付けに基づいている。 SAXSでは、これにX線誘起凝集を防止する最適化が含まれている。(トラブルシューティングを参照)。 SANSには、SANSデータを収集するのに必要な時間にわたるサンプルの安定性を評価することが含まれる。 SAXSとSANSの両方について、サンプルの取り扱い、調整、保管、および必要に応じて遠方の施設へサンプルを出荷することなどの物理的側面を考慮する必要がある。例えば、結晶をクライオ低温で保存することができるX線結晶学とは異なり、サンプルを凍結解凍する、または気泡を多量に導入するという単純な行為は、感知できるレベルの凝集を引き起こし、散乱データの解釈を台無しにする。したがって、サンプルの純度と安定性の両方を評価する必要がある。 クーマシーブルーで染色されたポリアクリルアミドゲル電気泳動(PAGE)ゲルは、タンパク質試料の精製純度を推定するためにほぼ普遍的に使用されている(PAGE - オプションA)。タンパク質の品質は、サイズ排除クロマトグラフィー(SEC - オプションB)または動的光散乱/静的光散乱(DLS / SLS - オプションC)によっても評価することができる。
オプションA:ポリアクリルアミドゲル電気泳動で試料純度を評価する。¶
- SASサンプルの調製には、ドデシル硫酸ナトリウムPAGE(SDS-PAGE)を使用してコンタミの存在を確認する。 特にコンタミが目的タンパク質よりも高い分子量を有する場合 さらなる精製工程(例えば、SEC)を導入する(図3)。 これらの高分子量コンタミは除去されなくてはならない。なぜなら、散乱強度は、分子の体積二乗に 比例するからである(式1)。高分子量のコンタミが存在せず95%以上に精製された試料ほとんどのSAS実験には十分であるはずである。
ご用心
SDS-PAGEゲル上の単一バンドは、必ずしもサンプルが溶液中で単分散であることを意味するものではない。 さらなる特性評価ステップ、例えばNative-PAGEおよびSECが必要である。
- SDSなしで実行するnative PAGEを実行して、タンパク質サンプルが主に均一であるか、または一連の会合種が広がっているかに関する詳細情報を取得する (図4)。
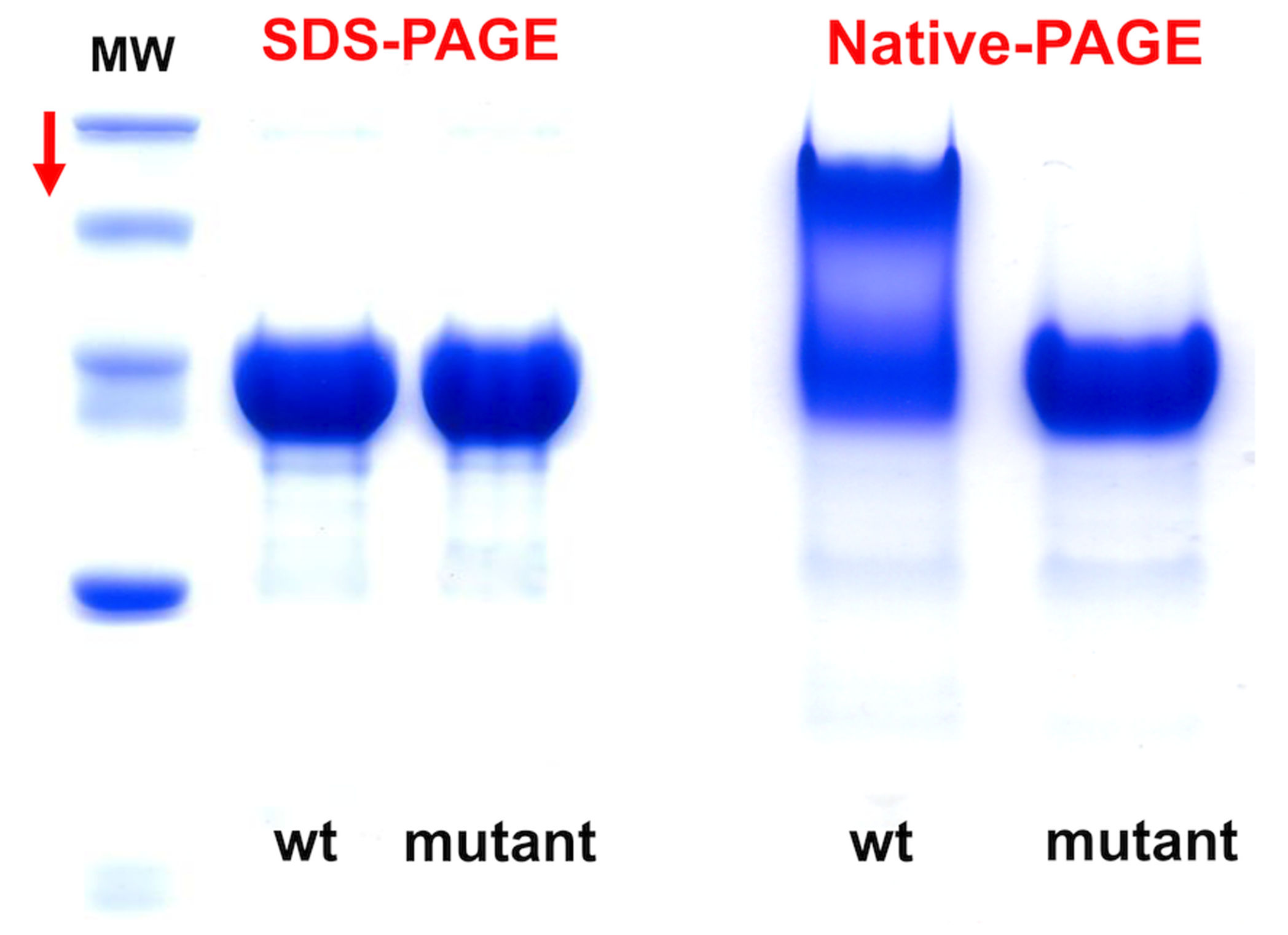
Native PAGEの価値 2つのタンパク質、野生型(wt)および突然変異体を、それぞれSDS-PAGEおよびNative PAGEを用いて分析する。 Native PAGEの結果は、突然変異体がタンパク質の会合状態を劇的に変化させることを示している。
- タンパク質は還元環境(例えば細胞内環境)においてしばしば発現されるが、ジスルフィド結合による会合を評価するために還元剤の有無による SDS PAGE、Native PAGEの結果を比較する。(還元剤は、例えば、ジチオスレイトール、DTT;β-メルカプトエタノール、βME;またはトリス (2-カルボキシエチル)ホスフィン-HCl、TCEP-HClである。 )。 必要であれば、還元状態(すなわち、分子間架橋がない)で目的のタンパク質を 維持するのに必要な還元剤濃度(典型的には1〜10mM)を決定する。
ご用心
タンパク質サンプルの熱変性を防ぐために、Native PAGEゲルが冷却されていることを確認すること。例えば、低温室で行う。
オプションB:SECを使用してサンプル品質を評価する。¶
試料中に存在する成分の正確な定量分析のために、紫外線(UV)分光法、可能であれば角度分散レーザー光散乱(MALLS)または 直角レーザ光散乱(RALLS)および屈折率(RI)測定と組み合わせて使用する、サイズ排除クロマトグラフィー(SEC)の価値は誇張ではなく重要である。 大部分の構造生物学研究室は、標準的な精製手順の一部としてSEC-UV装置を利用し、典型的に280nmでSEC溶出プロファイルを監視する。 しかし、SEC-RALLS-RIまたはSEC-MALLS-RI-DLS機器(Wyatt TechnologyのDAWN®HELIOSTM II + WyattQELSTMまたは Malvern InstrumentのOmnisec RevealおよびZetasizerμV)は、サンプル成分の完全な分析特性 (分子量の検量およびサイズ分析と組み合わせた連続流成分分離)ができるという点において重要となりつつある。
- SEC-UVを使用して、サンプルの濃度または時間依存性の安定性を、凝集体、高分子量オリゴマーなどの形成をモニターすることによって評価する
- SECを使用して、会合状態または試料成分(例えば、複合体または集合体)間の濃度依存的会合を調査する。これを行うには、少量のサンプル体積 (50〜100μl)を用いて、一連の濃度希釈をして、例えば、Superdex 200 Increase10/300 GLカラム(GE Healthcare Life Sciences)を使用する。 UV分光光度法を用いて、カラムにロードする濃度を変えてSEC溶出プロフィールの変化をモニターする。最も高濃度サンプルの時は、カラム容量に対して 過負荷にならないように注意すること。(分解能の喪失を引き起こす)。
- 完全に(またはほぼ完全に)形成された複合体を分離するのに必要なカラムにロードするサンプル濃度を決める。もし、タンパク質-DNA複合体に対して SEC-UVを行う場合、2つの波長、例えば280nm と260nmでのUV吸収をモニターすることが重要である。それにより、あるタンパク質複合体が形成され、 カラムを流れるとき間安定していることを示すことができる。
- (オプション)MALLSまたはRALLSと組み合わされたSEC-UV(RI)を用いて複合体の分子量(MW)および 結合化学量論を評価する。 このステップはSAXSサンプルの解析が困難な場合不可欠である(例えばバチルス・ズブチリス(Bacillus subtilis)由来のSdaタンパク質の分析を参照のこと)。
- SECから得られた結果を組み合わせて、PAGEの結果を解釈するのに役立てる。例えば、図8では、あるタンパク質がSDS-PAGEによると高純度で単分散 であるように見える。 しかし、SEC溶出UVプロファイルは、タンパク質はすでに自己会合の影響を受けていることを示している。
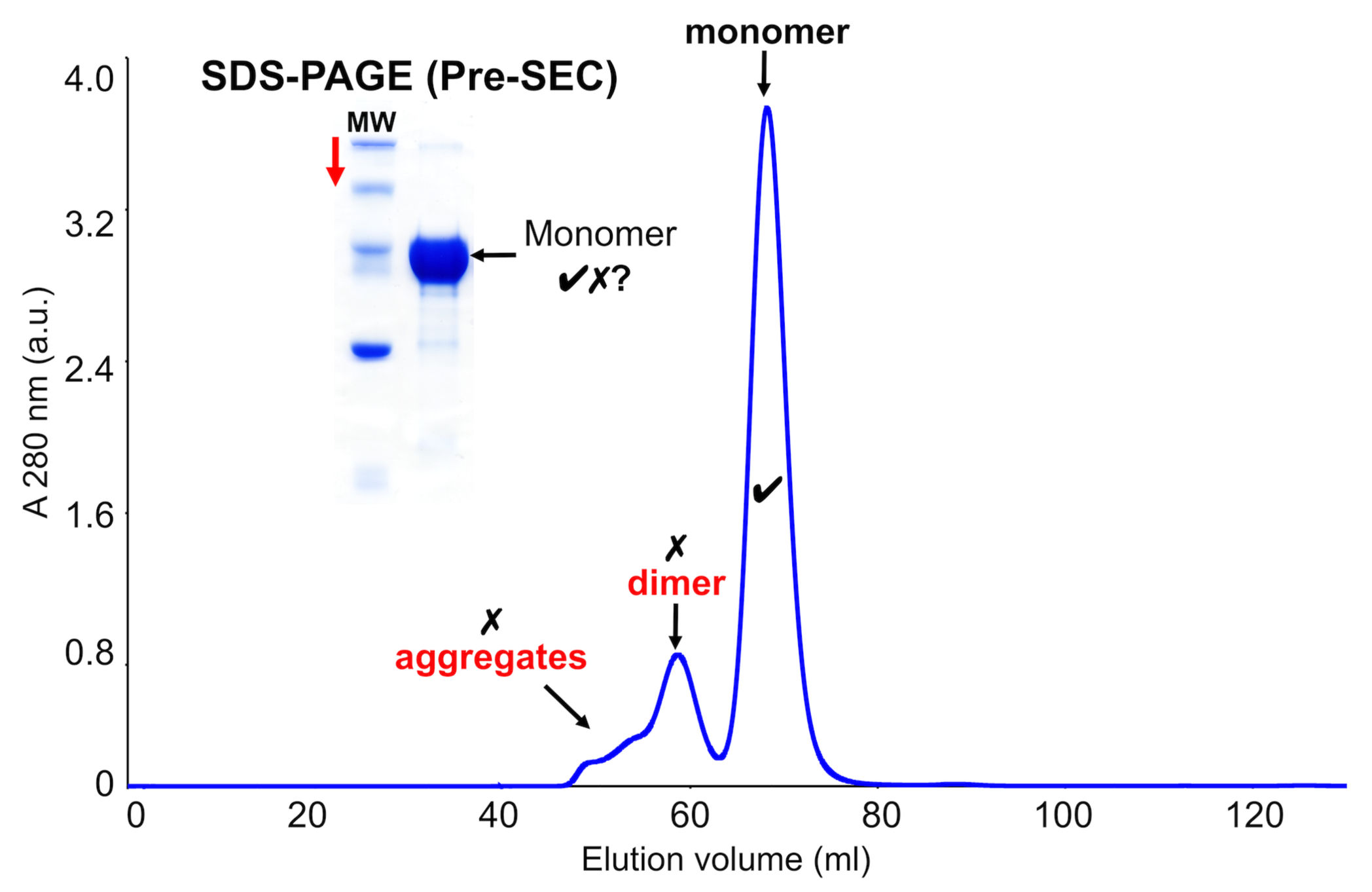
サンプルの特性評価:SECと組み合わせたSDS-PAGE 。 SDS-PAGEの結果は、タンパク質試料が十分純粋であることを示唆している。 しかし、SDS-PAGEの結果は、さらなるサンプルの特徴付けによる 裏付けがされなければ誤解を招く可能性がある。 SECは、サンプルが、自己会合凝集体、二量体および単量体を含む異種の粒子集団からなることを示している。
オプションC:DLS/SLSを使用してサンプル品質を評価する。¶
スタンドアローンのDLSとSLS装置(Wyatt TechnologyのDynaPro NanoStarTMやMalvern InstrumentのZetasizer Nano Rangeなど)は、数多くのサンプル環境 (pHやイオン強度の変化など)をすばやくスクリーニングし、サンプルの完全性(すなわち凝集物の形成など) を評価できる。 例えば、DLS / SLSは、 金属イオンおよび補因子物質等の添加の影響、還元剤、抗酸化剤(例えば、アスコルビン酸ナトリウム)または放射線損傷の低減のための安定化小分子 (例えば、5〜10 % v/v グリセロール)の添加の影響(トラブルシューティングを参照)を評価することができる。 SECベースのMALLS-DLSシステムと 比較してスタンドアローンDLS / SLSの利点は、非常に少量のサンプルを使用して分析を実行でき、数分で測定を完了できることである。
- 最小量のサンプル(2〜10μl)に関し動的光散乱を使用して多分散性を評価し、生体高分子の流体力学的半径( Rh )を測定する。 (正確な濃度推定と組み合わせて)SLSを使用して分子量とサンプル環境変化に対し分子量がどのように変化するかを評価する。 特に、DLSはサンプル中の凝集物の存在に非常に敏感で、その存在はSAS実験から得られた結果に対しても悪影響を与える。 一般的な経験則では、DLSで凝集体が検出されなかった場合、サンプルはSASに対して十分に高品質である。
- 様々なサンプル濃度、温度、時間の経過による凝集物の形成を監視するためにDLS/SLSパラメータによる評価を行う。この分析はサンプルが低収量、 生産が困難、または高価である時、単分散試料を調製し、単離するのに重要である。
- <オプション> SAXS測定後のDLSデータの Rh は、後続の形状解析に使用することができる。形状因子 \(Rg/Rh\) は、( Rg はSASから得られる) 一粒子内の電子密度分布を評価するためのパラメータを与える。( \(Rg/Rh\) ;球=0.774;フレキシブルコイル= 0.816;ロッド= 1.732)。 さらに、SLSから得られたMWは、SAXSから得られた分子量(MW)を検証するために使用でる。
DLSを使用したサンプル条件の最適化例¶
- 2×タンパク質ストック溶液(例えば、1〜10 mg.ml-1)を選択した緩衝液中に溶解する。
- 1つのタンパク質サンプルに、15μlの同じ緩衝液(すなわち、添加物を含まない)を加えてコントロールとする。 残りの4つのサンプルに、様々な濃度の添加剤を含む15μlのバッファーを加える(1, 2, 4, 8 ×目的最終濃度)。 例えば、50〜300mMのNaCl濃度をスクリーニングする。
- 泡立てないように、サンプルを注意深く混合し、高速遠心処理を5分間行う(例えば、マイクロ遠心分離機16000×gを使用)。 DLS / SLS測定のために注意深く、5μlのサンプルを取る。 SLSについては、濃度を記録する(手順2参照)。
- 各サンプルのDLS測定値から測定された多分散性および Rh パラメータを比較する。 分子量の変化を評価する。 溶液中の凝集体形成に添加剤の臨界閾値が存在するかどうかを評価する。
- 残りの10μlのサンプルを経時的に(例えば、3〜5日)保存し、DLS / SLS測定を繰り返し時間安定性と添加剤の有無による凝集状態を確認する。
DLSを使用した凍結融解の際の凝集体形成のチェック¶
DLSを使用して異なる凍結融解手順でサンプルの凝集状態がどうなるかを試験する。この例では、液体窒素中でサンプルを急速凍結し、 -80℃で保存することが記載されているが、ドライアイスで急速凍結または-20℃でゆっくり凍結保存した(推奨しません)サンプルを用いて 同様の試験を行うことができる
- 液体窒素を用いて2つのサンプル(例えば、エッペンドルフチューブ中で100μl)を急速凍結し、-80℃で保存する。 また、急速凍結をしていない(例えば、4℃で液体として保存する)サンプルを取り分けておく。
- サンプルの1つを急速解凍する(例:指先で慎重に)。
- 2番目のサンプルを氷上でゆっくりと解凍する。
- (例えば、凝集体の形成)などの変化を異なる凍結融解手順のサンプルと凍結解凍されていないサンプルとで比較する。
- 質問に答えてください:そもそもサンプルを凍結する必要がありますか?
- <オプション>さらなるSEC分析とPAGE分析を使用して、サンプルの凝集状態に関する異なる凍結融解手順の影響を調べる。
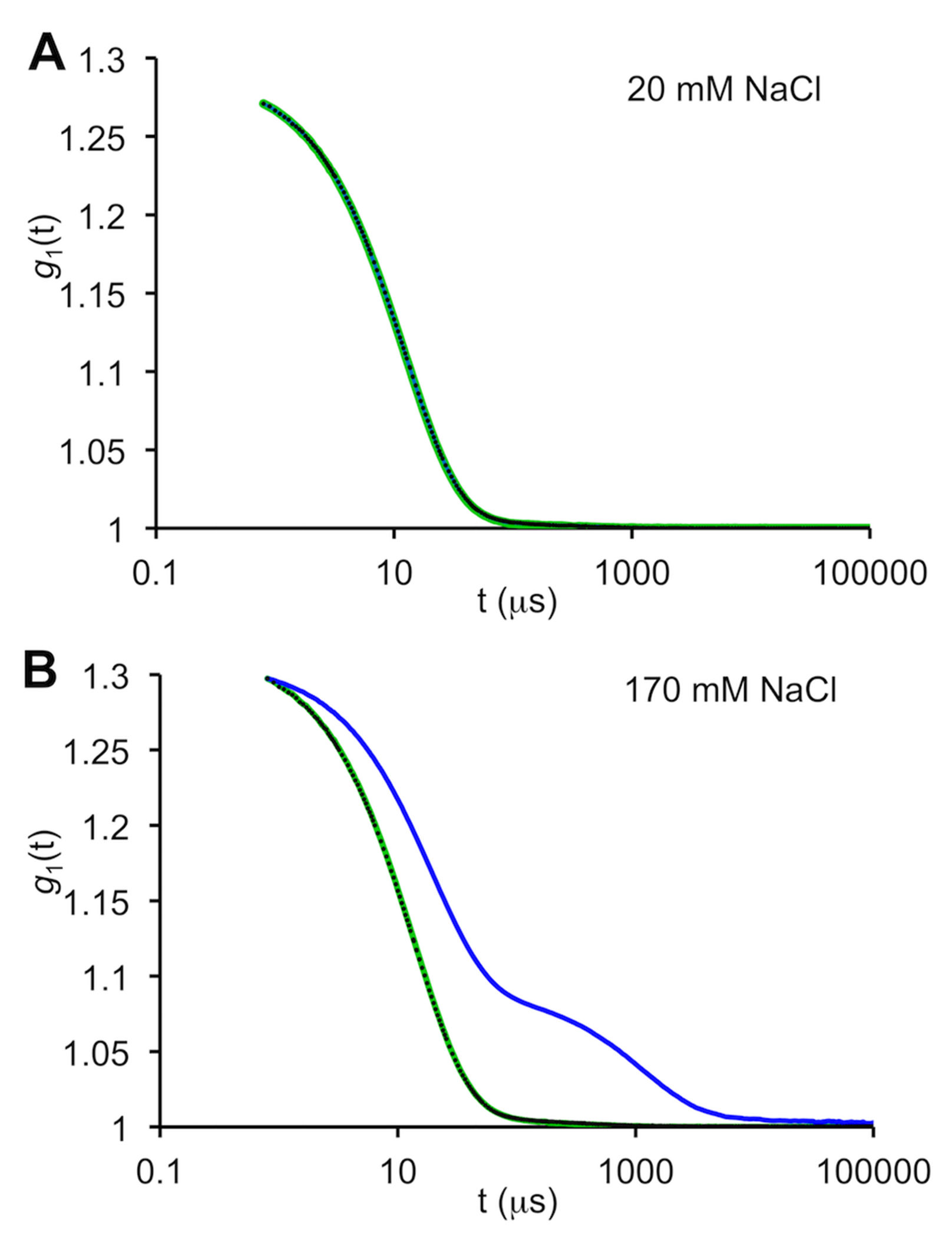
サンプルとサンプルの処理を特徴付けるツールとしてのDLS。 A.低塩緩衝液(20mM NaCl、40mM 酢酸ナトリウム、pH3.8)中のリゾチーム(9.1mg.ml-1)のDLS自己相関関数。 調製した直後の試料(緑色)を、 i. 液体窒素中で急速凍結し、迅速に融解(黒色点) ii. 瞬間凍結サンプルを氷上でゆっくりと解凍(青い線、不明瞭) した試料とで比較する。 自己相関データの指数関数的減衰とベースラインへのスムーズな戻りは、 3つのサンプルすべてが、異なる方法で保存、取り扱われても、凝集の影響を受けないことを示す。 B. NaCl濃度を170mMに増加させることは、新鮮かつ急速解凍されたサンプルの品質にほとんど影響しない。 しかし、急速凍結リゾチームを高塩濃度緩衝液中で氷上でゆっくり解凍すると、凝集物が生成される(青色)。 Wyatt TechnologyのDynaPro(登録商標)NanoStar(商標)機器を用いてデータを収集した。
サンプルが濃度または時間の影響を受けるかどうかを決定する。¶
サンプルの凝集が出現し始める(例えば、凍結融解および保存の有無にかかわらず)濃度閾値にサンプルが達しているかどうかを決定する。 DLS、PAGEまたはSECを使用してこの分析を行うことができる。サンプル濃度を上げるとSASデータにおけるS/N比を改善することができるが (下記式参照)、
\[I(q)=N(\Delta\rho V)^2P(q)\]粒子間相互作用の影響、特に濃度依存性の凝集体の形成をもたらすものを避けるにはSAS実験に おいて低濃度のサンプルを使用する必要があるかもしれない。
サンプルが時間的に安定しているかどうか、すなわち凝集またはより小さな成分への分解(例えば、加水分解、タンパク質分解など) に影響されやすいかどうか決定する。この分析には、DLS、PAGEまたはSECを使用できる。異なる温度での時間/貯蔵安定性を評価することは 遠方の施設にサンプルを運ぶ方法を決めるときに重要であろう(例えば、ドライアイスで凍結するか保冷剤の上で凍らせないでおくか)。
高分子が徐々に貯蔵管の側面に付着するかどうかを決定する。タンパク質の場合、これは、PAGEと並行して経時的に試料の濃度を評価する ことによってモニターすることができる。(濃度またはPAGEバンド強度の減少がポイント)。
濃縮、経時変化または凍結融解などによる凝集体が高速遠心分離(例えば、マイクロ遠心機16000gまたは30,000×g以上の超遠心分離機)、 希釈または遠心フィルターユニットによるスピン濾過(例えば、0.1-0.45μmの孔径のフィルター膜を使用)で除去できるか決定する。 そうでなければ、混入物を除去するためにSECステップをSAS実験の直前に追加する必要がある(例えば、小さな Superdex 200 Increase 5/150 GLカラム、GEヘルスケアライフサイエンス)。
ご用心
大きな凝集体や微粒子を除去するために使用される遠心フィルターの膜素材は、polyethersulfone(PES)、ポリフッ化ビニリデン(PVDF)誘導体、 ポリテトラフルオロエチレン(PTFE)、酢酸セルロース(CA)および硝酸セルロース(CN)などが使用できる。もし、高分子サンプルが 「不思議に消える」、または遠心フィルター後に凝集が増大するようになれば、意図と反してフィルター膜はサンプルと相互作用している。 必要に応じて、0.1〜0.45μm孔の異なる膜素材をテストしなさい。
ステップ2:サンプルと同一なバックグラウンド溶媒を得ること¶
高分子溶液散乱の場合、バックグラウンド溶媒による散乱の寄与をサンプルの散乱から差し引いて、目的の高分子または複合体からの散乱を得ることが非常に重要である。溶媒差分の誤差は、差分された散乱プロファイル中では残留溶媒項として残り(式1)、データから導出された構造パラメータに影響を与え得る(図7)。したがって、サンプルの溶液と同一に適合させた溶媒ブランクを得ることが不可欠である(序文参照)。ポイントは、大部分の散乱実験では、バックグラウンド溶媒の調製は、サンプルを調製することとほぼ同じくらい重要である。同一な溶媒の調製は、サンプル透析(オプションA)、サイズ排除クロマトグラフィー(オプションB)、または場合によっては分子量カットオフ遠心フィルター(オプションC)の注意深い適用によって達成することができる。
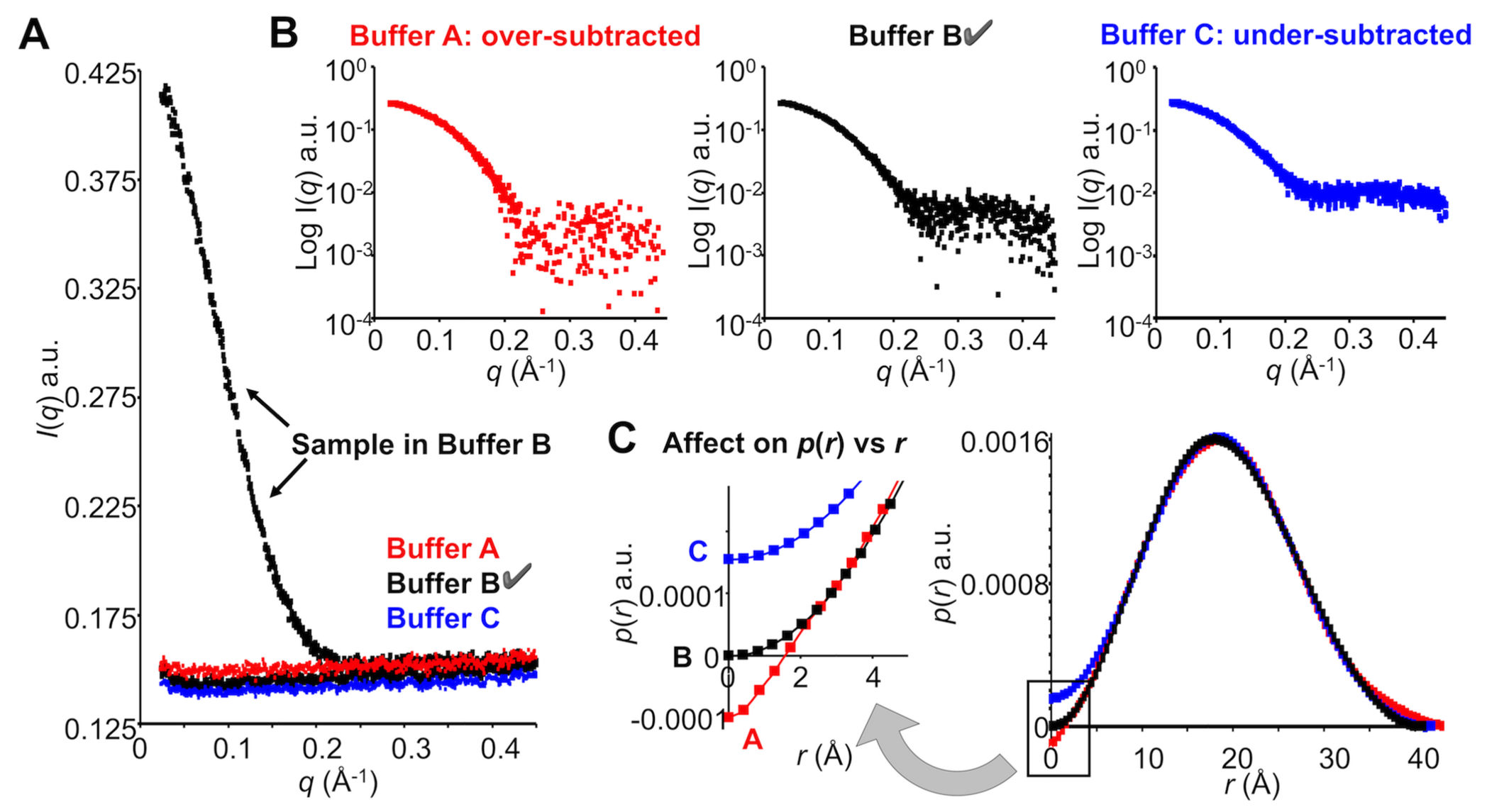
サンプル溶媒と同一な溶媒を得ることの重要性。 緩衝液B中の純粋で単分散のタンパク質サンプルとバックグラウンド差分の前の3種のバックグラウンド溶媒、A(赤色)、B(黒色)およびC(青色) から収集したSAXSデータ。緩衝液Bは透析を使用してサンプルと適合させたが、緩衝液Aは、誤ってラベル付けしたチューブ由来のものであり、 緩衝液Cは、冷蔵庫に保存した古い緩衝液から得た。 B.各緩衝液について、測定された平坦部の溶媒散乱強度は同様に見えるが、溶媒適合緩衝液Bのみが溶媒差分後にはじめてタンパク質からの正確な散乱 となることが明らかである。 C.溶媒差分が正しくないと、 \(p(r)\) 対 r のモデリングに影響が出る可能性がある。正しい溶媒適合バックグラウンド(黒色)は、 \(p(r)\) プロファイルを 原点で交差させ、過小差分バッファおよび過剰差分バッファはそれぞれr = 0で \(p(r)\) の正および負の値をもたらす。 [実際の実験データは、タンパク質、緩衝液Bおよび緩衝液Aについて提示されているが、緩衝液Cは説明のために調整した。]
ご用心
コントラストマッチングの溶媒を選択する際は、サンプルの濃度を考慮する必要がある。というのは、散乱強度は溶液中の均一な粒子の自乗に比例するからである (式2)。 透析は試料濃度をより制御しやすいが、一方、SECはサンプルがカラムを通過する際に希釈効果を与える。 高輝度放射光光源の場合、サンプルの希釈は問題では ないかもしれないが、実験室ベースの光源では、SEC中のサンプルの過希釈はデータのS/N比の低下を招き、より長い時間露光することが必要となる (試料は放射線損傷および時間安定性の両方が必要となる)。 希釈問題を解決するために、濃縮したサンプルをSECカラムにロードする (例えば、5〜15mg / ml)。 しかし、これは、サンプルを濃縮しても凝集が起こらず、カラムへの過負荷が生じてSEC分解能が失われないという前提に基づいて いる。
ご用心
溶媒ブランクを調製する際、サンプルの透析またはSECを単にスキップするため、サンプルの条件に「十分に近い」新しい溶液の成分を計量して作成したくなる。 このショートカットはほとんどうまくいかない。 透析またはSECを用いた溶媒交換する以外に散乱実験のためのサンプル溶媒条件(および特にその密度および吸収) を再現することは困難である。
オプションA:透析を行い、サンプルと適合する溶媒ブランクを得る。¶
- SAXS - とくにSANS - では、選択した溶媒に対し一晩サンプルの透析を行うこと。その際、目に見える気泡や空気ポケットが透析バッグ、ボタン またはカセットから除去されていることを確認する。
ご用心
透析を使用して溶媒マッチングを行うことは、場合によっては、実用的ではないこともある。例えばサンプルが時間の経過とともにゆっくりと自己凝集する場合。 透析処置の間に時間依存的な凝集によってサンプルが影響を受けないことを、DLSまたはSECを用いてテストすることは必要かもしれない。
透析したサンプルと透析後の両方から散乱データを収集する 透析後緩衝液は、SAXSまたはSANS測定で適合した溶媒ブランクとして使えるであろう。
SAXSの場合は、透析後バッファーを使用してサンプルを希釈し、濃度シリーズ(例えば、1、0.75,0.5および0.25x)を作成し、 濃度依存性粒子間干渉効果、 \(S(q)\) 、または、複合体の解離の評価を行う。
(オプション)SAXS由来の結果は、SANSのためのサンプル濃度の選択に役立てられるであろう(手順3、手順2を参照)。
オプションB:サンプル溶媒と一致した溶媒ブランクを得るためにSECを行う。¶
- SECカラムから溶出した目的のサンプルに対応する画分を集める。
- カラムを通過した緩衝液の少量(例えば、500μl)を収集してSAS実験のための一致した溶媒ブランクとする。 サンプル溶出ピークに可能な限り近い溶媒画分を収集する。
ご用心
SECカラムを通過したSECランニングバッファのみを使い、ストックボトルからのバッファは使わないこと。限られた量の小分子分画は カラム通過中の溶質/溶媒/カラムマトリックス相互作用によって引き起こされる。 この分画は、カラムを通過しなかった緩衝液と比較して、サンプルとともにカラムを通過した緩衝液の散乱長密度 および吸収特性を変化させる可能性がある。
- (オプション)高価で不安定な補因子(例えば、NADPH)をサンプルに添加する必要があるかもしれない。この場合、何リットルもの サンプル透析液またはSEC緩衝液の調製で、単純に浪費することはできない。 この場合の最善の方法、そしてサンプルを過度に希釈することを 回避する方法は、既に透析交換されたサンプル溶媒ブランク中に(10-50倍)の濃縮添加剤原液を少量加える。少量の等量の体積(または質量) の添加剤濃縮液をSASの直前にサンプルと溶媒ブランクの両方に正確に加えます。もし同量の質量で加える場合にはマイクロバランスを使用すること。
- (オプション)SAXSでは、SECとSAXSの測定値を直接組み合わせる。つまり、SECカラムから溶出した直後の分離されたサンプル分画からの 散乱データとともにSECカラムから溶出した溶出バッファーからの散乱データを収集すること。 ボックス1を参照のこと。
オプションC:遠心フィルターの使用(細心の注意を払って)。¶
透析またはSECの代替法は、遠心分離ベースの分子量カットオフフィルターを用いてサンプルを濃縮し、透過液を即席的にサンプルブランクとして 使用することである。 この手法は、注意深く行われるなら機能する。
ご用心
遠心濃縮器は、時にはかなりの量の小分子を膜に保持するので、透過液溶媒組成と比較してサンプルの溶媒組成が微妙に変化し、その結果、 溶媒の不一致が生じる。 いくつかの膜フィルターは、防腐剤(例えば、アジド、グリセロール)をコーティングして製造されるので、 完全に洗い落とされなければ不要な小分子を試料に導入してしまうかもしれない。もっと悲惨なのは、濃縮操作中に濃度勾配が発生して、 フィルター膜界面でサンプルが凝集する可能性がある。
- 常に適切な分子量カットオフを備えた新しい遠心濃縮器を選択する。一般的な経験則では、最小分子量カットオフは少なくとも目的の 高分子の分子量よりも3〜5倍小さくする。たとえば、タンパク質が25kDaのモノマー MW を有する場合、 MW カットオフが5kDa以下の フィルターを使用する。
- 少量の体積の緩衝液で注意深くフィルターデバイスの膜を洗浄する。(例えば、膜上を上下にピペッティングする)これにより、 製造プロセスから残っている小さな分子が除去される。余分なバッファーを取り除いてからサンプルをロードする。
- サンプルをカタログの指示通りの遠心速度または×gで遠心分離する。1回の長時間の連続遠心(例えば、20分間)よりは、 短時間で複数回の遠心(例えば、10×2分)を用い、間でサンプルを注意深く撹拌することが最良である。これは濃度勾配を防ぐのに役立ち、 ひいては膜/試料界面でのサンプル凝集の可能性を減少させる。各遠心の間に溶液を泡立てないように上下に注意深くピペッティングする。
- サンプルが所望の濃度に達したら、サンプルおよび膜を通過したバッファーを回収する。 回収したバッファーはSAXS実験のための溶媒ブランク として使用する。
ご用心
遠心濃縮中に溶液中のサンプルの濃度が減少するか、または一定となる(すなわち、増加しない)場合、これは タンパク質が膜フィルターと結合しているサインで、おそらく不可逆的な凝集物を生成するだろう(これはDLSでチェックできる)。 不可逆的な結合/サンプル凝集の可能性を減らすために、異なる種類の膜材料を試験することが必要な場合がある。 膜フィルターは、 再生セルロース(例えば、Amicon(登録商標)Ultra、Millipore; PierceTM Protein Concentrator)、ポリエーテルスルホン (PALL CorporationのNanosep(登録商標)およびMicrosep TM; GE Healthcare Life SciencesのVivaspin; Corning Spin(登録商標-X およびPierce PES Protein 濃縮器)ならびに改質ナイロンまたは親水性ポリプロピレン(Nanosep MF(登録商標)、PALL Corporation) が挙げられる。
注釈
手順1のタイムチャート。
SASのための精製された生体高分子の調整および特性評価は、数日から数週間に及ぶことがある。 SDS-PAGE分析は、セットアップ、ゲルの走行、 染色、および脱色を含みゲル当たり1〜2時間かかる。 Native PAGEのためには、走行するのにより長い最大4時間などの時間を必要とする。 SEC、SEC-MALLSまたはSEC-RALLSは、緩衝液の調製、カラムの平衡化、機器の較正、サンプルの走行などを完了するまでに日中7,8時間を要する。 そこで、カラムと検出器を一晩平衡させることで時間を節約しよう。 SEC-SAXSについては、ボックス1を参照のこと。スタンドアローンのDLSおよびSLSは、 30分間の機器平衡時間を必要とし、各測定は約100秒(例えば、それぞれ10×10秒)かかる。 サンプルテストに必要なDLSを実行するために 1日は必要。 サンプルの透析は、標準的には、1時間のセットアップ時間および一晩の緩衝液交換が必要である。 遠心に基づくタンパク質濃縮は、 完了に30〜60分かかることがあるが、遠心速度とサンプルの最終所望濃度に依存する。