次のページを参考に,ChemSketchで作成した分子構造をantechamberでpdbに変換し,packmolでbox内に配置しようとしてハマった点をメモ書き
https://makoto-yoneya.github.io/LAMMPS-organics/- lammpsはlinuxマシンで使用するのでインストールする必要はない
- ChemSketch, VMDはWindows版をインストール
- Ubuntu上ではgfortranとantechamber, packmolをインストールして使用する
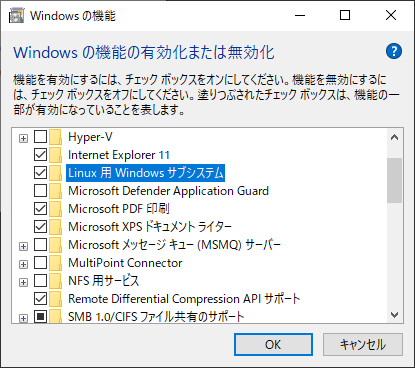
まずWSL(Windows Subsystem for Linux)を有効化する.次のページを参考に
https://www.pc-koubou.jp/magazine/21475
ただし,名称は以下のように変わっている点に注意
2021.03.18 追記
https://makoto-yoneya.github.io/LAMMPS-organics/の米谷様から
liblapack.so.3のエラーについて以下のコメントをいただきましたので,まずこちらを実行してからやれば以下のエラーは回避できるそうです.
|
エラーメッセージ(libgfortran.so.5, python3, liblapack.so.3)の件ですが、 https://makoto-yoneya.github.io/LAMMPS-organics/ のWSLのインストールに関する記述にある、 sudo apt install dos2unix python3-pip libgfortran4 libgfortran5 liblapack3 を実行してから手順を進めるとエラーとならない筈です(最近チェックしていませんが)。 (LammpsもWSLでaptでインストールされるものはUSER-MISCの問題は無い筈です)。 お役に立てば幸いです。 |
ubuntuをインストールした後,まずgfortranをインストールする. gfortranをインストールせずにpackmol, atomskをインストールしてpackmolを使おうとすると 次のようなエラーメッセージが出る
packmol: error while loading shared libraries: libgfortran.so.5: cannot open shared object file: No such file or directory |
sudo apt-get install gfortran |
wget https://github.com/makoto-yoneya/makoto-yoneya.github.io/raw/master/LAMMPS-organics/install_packmol-atomsk.sh sh install_packmol-atomsk.sh |
wget https://github.com/makoto-yoneya/makoto-yoneya.github.io/raw/master/LAMMPS-organics/install_WSLmisc.sh sh install_WSLmisc.sh |
ChemSketchで作成したmolファイルを「ドキュメント」フォルダに入れた場合は, WSLからは/mnt/c/Users/(windowsユーザー名)/documentsでアクセスできる.
手順は
- ChemSketchで基本分子鎖構造作成, hogehoge.molと保存
- WSLのUbuntuを立ち上げて,cd /mnt/c/Users/(windowsユーザー名)/(保存したディレクトリ)
- antechamber -fi mdl -i hogehoge.mol -fo pdb -o hogehoge.pdb -dr no としてmolをpdbに変換
- packmolのインプットファイルhogehoge.txtを作成し(参照ページ), packmol < hogehoge.txtとしてセル中に充填
- VMDで確認
次はmolファイルからlammps実行用のスクリプトを作成するためにmoltemplate, atomskを使うが, 書いてあるとおりにやってトラブったポイント
- moltemplateをインストール
wget https://github.com/makoto-yoneya/makoto-yoneya.github.io/raw/master/LAMMPS-organics/install_moltemplate.sh sh install_moltemplate.sh
- 書かれている通りにantechamberでmol2ファイルを作成
下はCPUメッセージantechamber -fi mdl -i noname01.mol -fo mol2 -o noname01.mol2 -at gaff -c gas -rn noname01 -dr no Welcome to antechamber 20.0: molecular input file processor.
- mol22lt.plでltファイル作成
$ mol22lt.pl < noname01.mol2 > noname01.lt
- moltemplate.shを書かれているように打ち込むと「pythonがない」とエラー
$ moltemplate.sh -atomstysle full -pdb pa-128.pdb system.txt moltemplate.sh v2.18.6 2020-8-13 Error: moltemplate.sh requires python
- 調べるとpython3はインストールされているので,シンボリックリンクを作成
$ sudo ln -s /usr/bin/python3 /usr/bin/python [sudo] password for mdcalc: mdcalc@YSR2019:/mnt/c/users/yashiro/documents$ which python /usr/bin/python
- 上のmoltemplate.shの文を再度実行.無事動き,大量のメッセージのあとにファイルが4つ作成される
lttree_postprocess.py v0.5.1 2017-8-23 lttree_postprocess.py: -- No errors detected. -- raw2data.py v0.44.0 2016-12-21 "Atoms" column format: atom-ID molecule-ID atom-type q x y z copied atomic coordinates into system.data postprocessing file "system.in.settings" mdcalc@YSR2019:/mnt/c/users/yashiro/documents$ ls system* system.data system.in system.in.init system.in.settings system.txt - この4つのファイルをlinuxマシンにコピーして,system.inを書き換えてエネルギー最小化のみさせようとすると次のエラーが.
mdcalc@univ2015-2 test]$ mpirun -np 8 lmp_mpi < system.in LAMMPS (11 Aug 2017) ERROR: Unknown dihedral style fourier (../force.cpp:537) Last command: dihedral_style hybrid fourier
- 調べるとUSER-MISCパッケージを入れてコンパイルしなければならないようで, lammpsのsrcディレクトリでmake yes-USER-MISCした後, make mpiでコンパイル (lammpsインストールのページを参照), できた実行ファイルを入れ替える(~/bin/lmp_mpiにおいてたのを/bin/lmp_mpiと間違って, 変えたつもりで変わってなくてしばらくトホホ道を彷徨った)
- 無事lammps動き,作成されたpa-128_min.lmpをatomskでpdbファイルにしようとしたら次のエラー(winmostarに移行したため放置)
$ atomsk pa-128_min.lmp pdb atomsk: error while loading shared libraries: liblapack.so.3: cannot open shared object file: No such file or directory
- moltemplateをインストール