intel parallel studioでVASP5.4.4.pl2とlammpsのコンパイル,CUDAセットアップ
インテル Parallel Studio XE 2020 Composer Edition for Fortran & C++ Linux アカデミック 日本語版+OpenMPI-4.0.5でVASP5.4.4.pl2のコンパイル
- プリインストール版計算機を買ったんだが環境変数が設定されてなく,ifortが使えなかった
→ログインディレクトリの.bashrcに以下を追加し,source ~/.bashrcして環境変数更新
source /opt/intel/bin/compilervars.sh intel64
- https://www.open-mpi.org/から安定版のファイルをダウンロード.このときはopenmpi-4.0.5.tar.gz
ホームフォルダにinstalled_packagesというディレクトリを作って,そこにこのファイルを置き,そのディレクトリで以下を実行
tar zxvf openmpi*.gz
- 展開されたフォルダに移動して以下を実行し自分の環境に合わせたコンパイル準備
./configure --prefix=/usr/local/openmpi CXX=icpc CC=icc FC=ifort F90=ifort F77=ifort
上ではインストールディレクトリを/usr/local/openmpiに,Cコンパイラをicc, C++コンパイラをicpc, fortranコンパイラをifortに
当然ながらicc, icpc, ifortと打って「コマンドが見つかりません」と出たらインテルコンパイラが入ってない
- make allと打って一般ユーザーでコンパイル
- エラーが出なければスーパーユーザーになってmake all install 2>&1 | tee make.log
(画面とmake.logにコンパイル時のメッセージ出力,下記のような警告は出てるがコンパイルは出来た)
../../../.././ompi/mpiext/pcollreq/use-mpi-f08/mpiext_pcollreq_usempif08.h(33): リマーク #5140: 識別できないディレクティブです。
!DIR$ IGNORE_TKR sendbuf, recvbuf
|
ハマるな危険!
スーパーユーザーになってインストールする際に,source /opt/intel/bin/compilervars.sh intel64を実行しないとエラー出る(当然)
ハマりましたw
- 一般ユーザーに戻って.bashrcに以下を追加
export MPIROOT=/usr/local/openmpi
export PATH=$MPIROOT/bin:$HOME/bin:$PATH
export LD_LIBRARY_PATH=$MPIROOT/lib:$LD_LIBRARY_PATH
- source ~/.bashrcして環境変数更新,mpif90と打って
「ifort: コマンドライン・エラー: ファイルが指定されていません。ヘルプを表示するに は "ifort -help" と入力してください。」
と出ること確認,以上でOpenMPIのセットアップ完了
- VASPのIDとPWを入手してダウンロードサイトから
vasp.5.4.4.pl2.tgz
をダウンロード,installed_packagesに保存し,そのディレクトリでtar zxvf vasp*.tgzとして展開
- archの中のmakefile.include.linux_intelをmakefile.includeとし,中身を編集
FC = mpif90
FCL = mpif90 -mkl=sequential -lstdc++
BLACS = -lmkl_blacs_openmpi_lp64
MPI_INC = /usr/local/openmpi/include
エラーがでなければ,binの下にvasp_gam vasp_ncl vasp_stdが生成される.これを実行パスに置いて使用する
BLACSを変えずにコンパイルすると,次のようなエラーで停止する(エラーメッセージがヒントに)
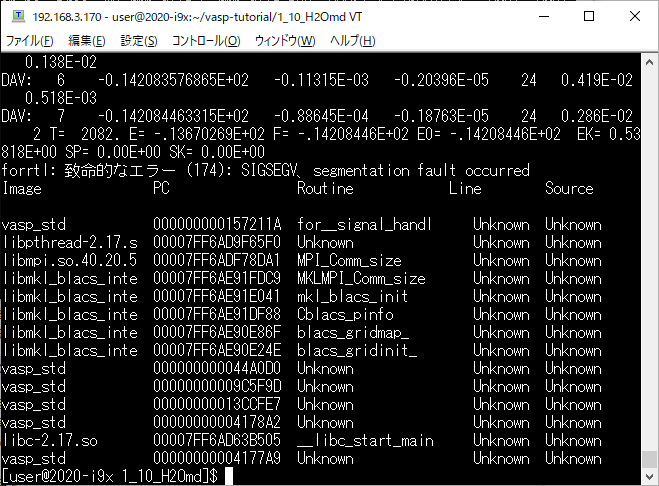
parallel_studio_xe_2019_update4_cluster_editionのインストール
- parallel_studio_xe_2019_update4_cluster_edition.tgzを入手
- インストール方法はググれば出てくるので省略
- 必要要件不足が表示されたので,
#yum install libXScrnSaver 実行
- ~/.bashrcに以下を追加
source /opt/intel/bin/compilervars.sh intel64
(実行出来ないときはsource ~/.bashrcしてみる)
→ifort, icc, mpiifort, mpiexecなどが実行出来るようになる
lammpsのコンパイル
- 最新版ではコンパイル失敗したが,lammps-11Aug17ではサクッと行けた
2020.10.01追記最新版(Mar20)はtar.gzとなっていながら中身は.tar, 下のCUDAセットアップ参照
- lammps-stable.tar.gzをフォルダにコピー
- $tar zxvf lammps*.gzで展開,lammps-11Aug17フォルダが生成される
- $cd lammps-11Aug17
$cd src
$make mpi
これでlmp_mpiが生成された
- lmp_mpiをコピーして使うか,実行パスが通ったフォルダにコピー
(~/.bashrcの中にexport PATH=$HOME/bin:$PATHと記述して~/binにlmp_mpiを置く)
- lammps-11Aug17/example/aireboに移動して
mpirun -np 4 lmp_mpi < ./in.airebo で動くこと確認
(注意点:< ./in.airebo とせずに < in.aireboとすると,binにあるlmp_mpiがbinにin.aireboを探すためファイルが無い,とエラー返す)
- 2019.07.10追記 過去の切断シミュレーションのスクリプト使ったら
ERROR: Unknown fix style rigid (../modify.cpp:854)
Last command: fix 2 wall rigid single force 1 off off off
とエラーが.lammpsコンパイル時にrigidパッケージをオンにしていなかったため.
- lammps/srcフォルダでmake package-statusと打つとパッケージが確認出来る
- make rigid yesでrigidパッケージを有効に
- make mpiで lmp_mpiを作り直す
CUDAのセットアップ
- ・cuda_9.2.148.396.37_linux.runをダウンロード
- スーパーユーザーになる
- 端末でinit 3と入力してXserver停止
- #sh cuda_9.2.148.396.37_linux.runを実行,デフォルトで進める
- init 5を実行してXserver起動
- ホームディレクトリの.bashrcに以下を追加
export PATH=/usr/local/cuda-9.2/bin${PATH:+:${PATH}}
export LD_LIBRARY_PATH=/usr/local/cuda-9.2/lib64${LD_LIBRARY_PATH:+:${LD_LIBRARY_PATH}}
|
GPUを使用するようにlammpsコンパイル
- 最新版lammpsをダウンロード,500MBぐらいあって時間かかる

- lammps-stable.tar.gzをlinuxのホームフォルダにコピー
- $tar xvf lammps*.gzで展開(zxvfするとエラー,.gzでありながらtarファイル),lammps-3Mar2フォルダが生成される
- $cd lammps-3Mar2
$cd lib/gpu
- Makefile.mpiをviエディタで編集.CUDA_HOME =/usr/local/cudaとなってるのをCUDA_HOME =/usr/local/cuda-9.2に変更
- make -f Makefile.mpiを実行
- 2020.10.08追記:CUDAインストール済みの計算機にてmake -f Makefile.mpiでエラーがでる.
CUDA_ARCH = -arch=sm_20を#つけてコメントアウトして,一番最新のCUDA_ARCH = -arch=sm_75を有効にしてコンパイル
このページでsm_75を調べた
- ./nvc_get_devicesとうつとGPU情報が表示される
- cd ../../srcしてlammps-3Mar2の下のsrcに移動
- make psしてオプション確認,GPU使用,剛体,Tersoff,剛体,xtcファイルdumpなどのオプションのため以下を実行してYESにする
make yes-gpu
make yes-rigid
make yes-peri
make yes-USER-MISC
make yes-USER-MOLFILE
make yes-MOLECULE
make yes-MANYBODY
make yes-MISC
make yes-KSPACE
|
- make mpiしてコンパイル,長々とメッセージ流れた後にエラーメッセージでなければlmp_mpiが生成される.
下はコンパイル成功したときのメッセージ
write_restart.o xdr_compat.o -lgpu -ldl -lcudart -lcuda -o ../lmp_mpi
size ../lmp_mpi
text data bss dec hex filename
14944214 22296 362296 15328806 e9e626 ../lmp_mpi
make[1]: ディレクトリ `/home/mdcalc/install_packages/lammps-3Mar20/src/Obj_mpi' から出ます
|
- 2020.10.08追記 ifort+OpenMPIでこちらのようなエラーでコンパイル失敗.
書かれているようにsrcディレクトリのpair_list.h, pair_list.cppを削除してコンパイル
- 実行ファイルlmp_mpiを実行パスを通したフォルダにコピー⇒$cp lmp_mpi ~/bin/lmp_mpi
- 実行パスを通すには.bashrcを編集
export PATH=$HOME/bin:$MPIROOT/bin:$PATH
|
上の例ではもともとの$PATHに,$MPIROOT/binと$HOME/binを追加している.