







研究内容
細胞中では、すべての分子の相互作用が同期して起こるわけではなく、ばらばらのタイミングで起こります。
さらに、個々の分子の相互作用時間は1秒以下ということも多いです。
相互作用する分子の割合が10%以下ということも多々あります。
そのため、細胞中の多数の分子の平均を計測していると、分子が“はたらく仕組み”を見出すことは極めて困難です。
そこで我々は、細胞中のタンパク質や脂質分子を1分子ずつ観察し(ときには、10,000フレーム/秒という世界最高速で!)、起きている事象の時間や頻度などの統計をとることで、分子のはたらく仕組みの解明を目指しています。
(もちろん、多数分子の同時観察時の平均値と1分子観察で得られた結果との整合性がなければなりません)
1. 高速超解像観察法による生細胞膜構造の動態の解明
PALM法やdSTORM法などの1分子観察をベースにした超解像顕微鏡法では、明滅する色素を1分子観察して、500-10,000フレームの画像中の輝点の位置を点描画を描くようにプロットして、1枚の超解像画像を得ます。
そのため、空間分解能は約20nmと高いのですが、非常に長い時間を要し、通常、固定細胞を観察します。
我々は、この問題を解決するために、高速で明滅する輝点を1分子観察することにより、1枚の超解像画像を短期間に取得し、得られた画像をつないで、動画を取得することに成功しました。
この手法により、生きている細胞の膜構造を20nmの空間分解能で変化する様子を追跡できるようになりました。
例えば、図1は細胞形質膜上のカベオラの超解像動画を5秒おきに切り出した画像ですが、矢尻先のカベオラは、隣の画像の同じ位置にはないので、この時間スケールで形成・消滅していることが分かります。

図1.5秒ごとに取得したPZ-HPV-7細胞上のカベオラのPALM画像(上)と通常の蛍光顕微鏡画像(下)。PALM画像中の黄色丸内にカベオラが2つずつあるが、通常法では1つしかないように見える。マゼンタ矢尻先にはカベオラがあるが、5秒前あるいは後で同じ場所にはカベオラがないことが分かる。
2. がん細胞由来の細胞外小胞による標的細胞の改変機構
近年、細胞間の情報伝達の担い手として、細胞外小胞が非常に注目されています。
細胞外小胞の中には、分泌した細胞由来の核酸やタンパク質などの情報がつまっており、これが標的細胞内に取り込まれることにより、標的細胞の性質を変えることができます。
例えば、がん細胞由来の細胞外小胞が他の臓器の標的細胞に取り込まれると、その細胞の周りにはがん細胞が転移しやすい環境が形成されると言われています。
しかし、細胞外小胞がどのようにして標的細胞に結合し、取り込まれるのか、その機構の多くは不明でした。
我々は、高速1分子・超解像顕微鏡技術を開発し、この問題に挑みました。
細胞外小胞にはサブタイプがあり、PC3細胞由来の細胞外小胞の場合、CD63だけを含有するものが60%、CD63, CD9, CD81のすべてを含有するものが40%あることが明らかになりました。
また、図2に示しますように、がん細胞由来の細胞外小胞中のインテグリンα6β1とα6β4が、標的細胞上の細胞外マトリックスのラミニンに結合することにより、細胞外小胞と標的細胞は結合することが明らかになりました。
がん細胞由来細胞外小胞中のラミニン受容体インテグリンα6は、4回膜貫通型タンパク質のCD151により、結合が促進されることも明らかになりました。
一方で、細胞外小胞内のタリンやキンドリンなどはインテグリンのラミニン結合活性を向上させないことも分かりました。
また、インテグリンとラミニン結合が主要な結合を担っていましたが、その他に、糖脂質ガングリオシドの一つのGM1もラミニンに結合することが明らかになりました(Isogai et al., J. Cell Biol., 2025)。
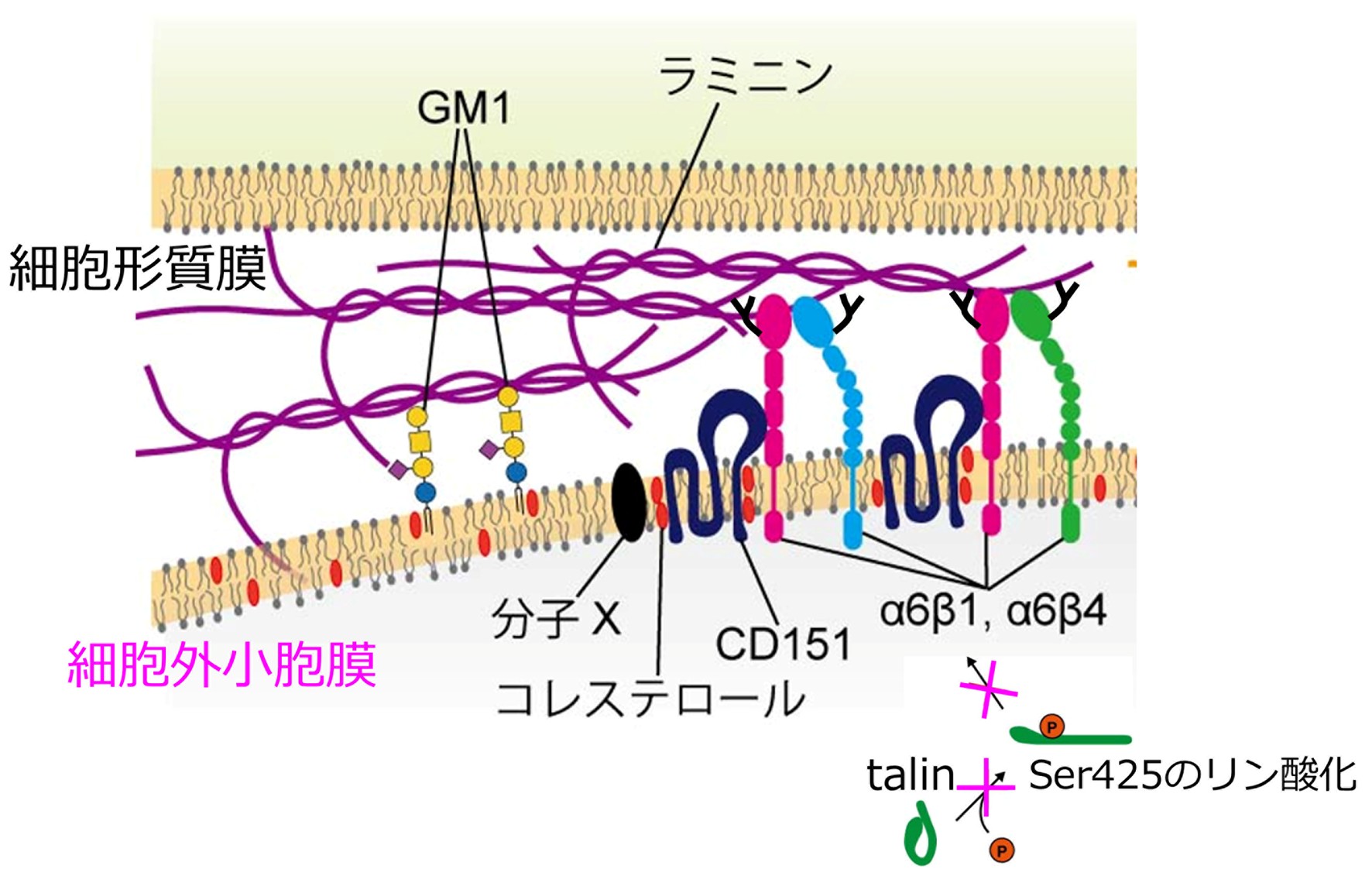
図2.細胞外小胞膜上のインテグリンα6β1, α6β4が、標的細胞膜上のラミニンと結合して、細胞外小胞は標的細胞に結合する。一方、ファイブロネクチン受容体は結合活性がない。テトラスパニンのCD151がインテグリンα6に結合し、ラミニン結合活性を向上させている。細胞外小胞中のtalinやkindlinはインテグリン活性化には貢献していない。コレステロールを介してラミニン受容体の阻害分子が結合している。細胞外小胞上のガングリオシドGM1もラミニン結合活性を有している。
次に、標的細胞による細胞外小胞の取り込み機構を解明するために、細胞外小胞をCD63などの4回膜貫通型タンパク質で蛍光標識して1粒子追跡しながら、取り込み膜構造を超解像動画観察しました(図3)。
結果、細胞外小胞の多くは、LAMP-2Cをマーカーとして観察したクラスリン非依存性エンドサイトーシス領域経由で取り込まれることが明らかになりました。
また、膜流動性が低いCD63含有細胞外小胞だけが、カベオラ経由でも取り込まれることが明らかになりました(Hirosawa et al., Nat. Commun., 2025)。
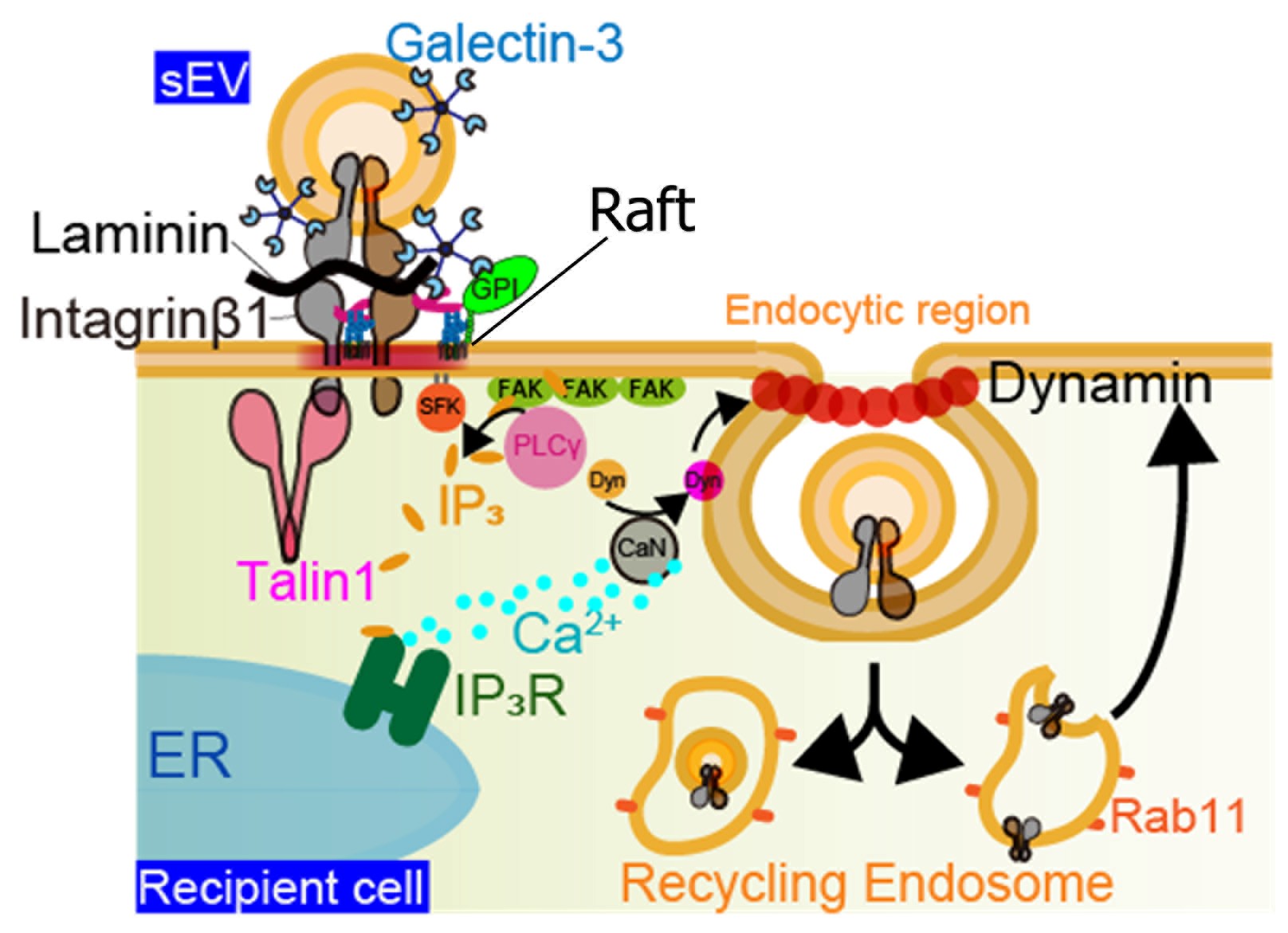
図3.細胞外小胞は、図2で示したように標的細胞上のラミニンに結合する。また、細胞外小胞上にはGalectin-3が被覆されていて、細胞膜上と細胞外小胞膜上のLacNAcを保持する膜タンパク質同志が架橋される。Lamp-2cも膜タンパク質の一つであり、細胞外小胞とLamp-2c複合体はクラスリン非依存性エンドサイトーシス経由で標的細胞により取り込まれる。この取り込みは、細胞外小胞の標的細胞膜への結合により促進される。細胞外小胞直下の標的細胞膜にインテグリンβ1が濃縮し、続いてTalin-1がクラスター形成し、Src family kinase(SFK)の活性化を誘起する。活性化SFKはFAKクラスターへのPLCγのリクルートを誘起し、PI(4,5)P2からIP3を産生させ、ER上のIP3受容体経由での細胞内カルシウム応答を誘起する。細胞内カルシウム上昇が、カルシニューリン経由でダイナミン活性を向上させ、最終的にクラスリン非依存性エンドサイトーシスやカベオラ経由での細胞外小胞の取り込みを促進する。
驚いたことに、細胞外小胞の結合シグナルが、標的細胞による細胞外小胞の取り込みを促進することが明らかになりました。
すなわち、細胞外小胞が標的細胞膜に結合すると、その直下にインテグリンβ1が濃縮され、タリンのクラスター形成を誘起しました。
その後、Src family kinase経由でPLCγが活性化され、標的細胞膜内層のPI(4,5)P2のIP3への分解が起き、ER上のIP3受容体に結合すると、細胞内カルシウム上昇が起きることを見出しました。
その後、カルシニューリン経由でダイナミン活性が向上し、細胞外小胞の取り込みが促進されることを明らかにしました。
興味深いことに、このシグナル伝達は、細胞外小胞を分泌した元のがん細胞に細胞外小胞が結合しても起きず、異種の細胞への結合後においてのみ起きることが明らかになりました (Hirosawa et al., Nat. Commun., 2025)。
細胞外小胞は、医療、創薬他、様々な領域で応用が期待されていますので、その動作原理を明らかにすることは極めて重要です。
今後も、先端イメージング技術を用いて、研究を進めてまいります。
プレスリリース2025 [Isogai et al., 2025 J. Cell Biol.]
プレスリリース2025(1) [Hirosawa et al., 2025 Nat Commun.]
プレスリリース2025(2)
プレスリリース2025(3)
3. 自然免疫応答分子STINGの活性化機構の解明
細胞がDNAウイルスに感染すると、それに応答して自然免疫応答、炎症が誘導されます。
STING経路はDNAウイルス感染から身体を守っていますが、一方、異常な活性化は自然免疫疾患、神経変性疾患、がんなど、様々な疾患を引き起こします。
私たちは、東北大学田口友彦研究室との共同研究で、STINGの活性化機構を1分子・超解像顕微鏡観察により解明しました(Kemmoku et al., Nat. Commun., 2024)。
STINGはパルミトイル化脂質修飾を介してトランスゴルジネットワーク(TGN)のコレステロールを含む脂質マイクロドメインで平均20分子以上の大きなクラスターを形成すること、下流のTBK1はSATINGの大きなクラスターが形成されて初めて、STINGへリクルートされることを発見しました(図4)。
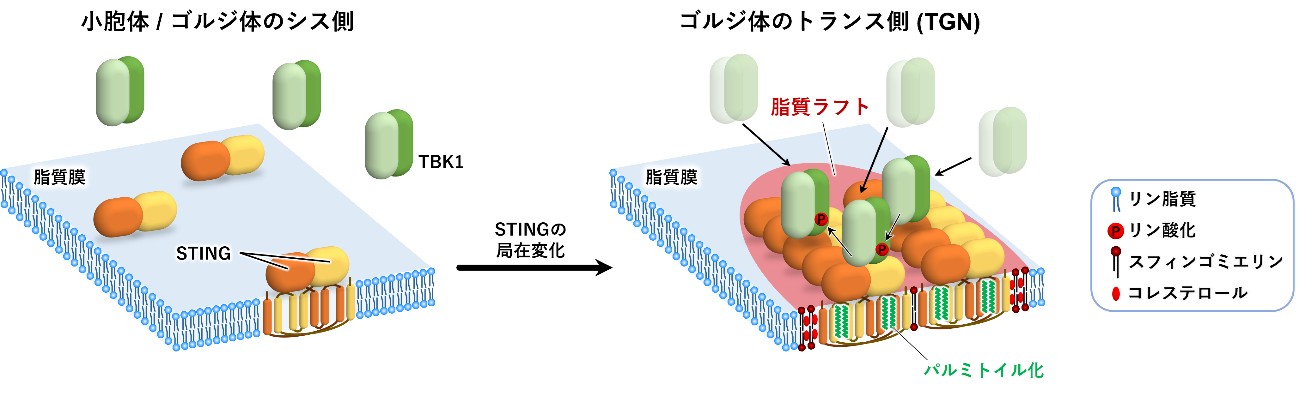
図4.刺激前STINGは小胞体上でダイマーとして存在する(左)。リガンド刺激後、STINGはトランスゴルジネットワークへ移動し、コレステロールやパルミトイル基などの間の脂質相互作用により、20分子以上からなる大きなクラスターを形成する。下流信号分子TBK1は分子密度が高い大きなSTINGクラスターへリクルートされる。
プレスリリース2024(1) [Kemmoku et al. 2025 Nat. Commun.]
プレスリリース2024(2)
4. 細胞膜構造、特に脂質ラフト構造とシグナル伝達機構の解明
細胞膜上の情報伝達のプラットフォームとしてラフトという概念が、Kai Simonsらにより提案されて(Simons and Ikonen, Nature, 1997)から久しいですが、
ラフトは光学顕微鏡でみることはできないほど小さいことや、分子の出入りが激しい(可塑性が高い)ために「ラフトとは何か?」いまだに議論が続いています。
初期のラフト研究では、界面活性剤を用いたり、免疫染色法が利用されていましたが、いずれもアーチファクトを誘導してしまうことが明らかになっています(Tanaka and Suzuki, Nature Methods, 2011)。
我々は、上記の問題を避けるために、できるだけ摂動を加えずにラフトを調べる必要があると考え、代表的なラフトマーカーであるGPI-アンカー型受容体(CD59やDAFなど)を生細胞膜上で1分子観察する実験手法を取ることにしました。
試行錯誤と様々な実験結果から、図1に示しますように、GPIアンカー型タンパク質は、まずは、タンパク質相互作用によりホモダイマー(同種の2量体)を形成し、続いて脂質相互作用がホモダイマーを安定化している(図1左)ことを見出しました。
このホモダイマーは、さらに大きな会合体のラフトを形成するための、最も基本的なユニットのひとつであると提案しています。現在、この説の検証を進めています。
さらに、細胞質側ドメインを持たないGPIアンカー型タンパク質が、どのようにして細胞の外から内へシグナル伝達を引き起こすのかについて研究を進めています。
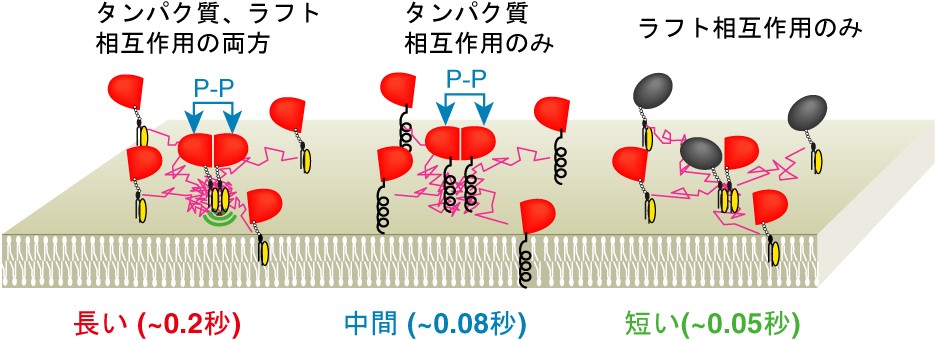
図5.GPIアンカー型タンパク質 CD59は、タンパク質相互作用によって二量体が誘起され、それはラフト相互作用により安定化された。タンパク質相互作用がなく、ラフト相互作用のみでは、寿命の長い二量体は形成されなかった。
プレスリリース2010 [Tanaka and Suzuki et al., 2010 Nat. Methods]
プレスリリース2012 [Suzuki et al., 2012 Nat. Chem. Biol.]
5. 細胞膜上での糖鎖動態の解明
極めて重要ですが、まだ不明な部分が多い「糖鎖」の細胞膜上での動態に注目し、その機構の解明を目指しています。
例えば、すでに、安藤先生、木曽先生のグループ(元京都大学物質細胞統合システム拠点サテライトラボ、現岐阜大学生命の鎖統合研究センター)との共同研究により、世界で初めて糖脂質のガングリオシド蛍光プローブ4種(図2)の開発に成功しました。
ガングリオシドは代表的な「脂質ラフト」のマーカーであると考えられています。
しかし、ラフトマーカーとなりうるガングリオシド蛍光プローブが存在していなかったため、今まで、ガングリオシドの生細胞膜のラフト中での動態はほとんど研究されていませんでした。
我々が開発したガングリオシド蛍光プローブは、ラフトマーカーとなりうる初めてのものとなりました。
ガングリオシド蛍光プローブの1分子ずつの挙動を観察した結果、ガングリオシドは、定常状態では極めて小さいラフトに非常に短期間出入りするのみですが、
GPIアンカー型受容体の2量体や、受容体をリガンド刺激後にできる安定化4量体ラフトには、より長期間滞在することを見出しました(図3)。
言い換えると、GPIアンカー型受容体は、その会合と同時に他のラフト親和性脂質をリクルートし、ラフトを安定化していることを見出しました。
また、このことは、大阪大学・村田研、九州大学・松森研との共同研究で開発しましたスフィンゴミエリン蛍光プローブを用いた実験でも裏付けることができました(Kinosita and Suzuki et al., JCB, 2017)。
以前の報告によりますと、ガングリオシドは、細胞膜上の受容体活性を制御していることがよく知られていますが、実際に生きている細胞膜上では、どのように制御しているのかは、ほとんど研究されていません。
現在、我々は、特に糖鎖相互作用に焦点を置き、この課題に挑戦しています。
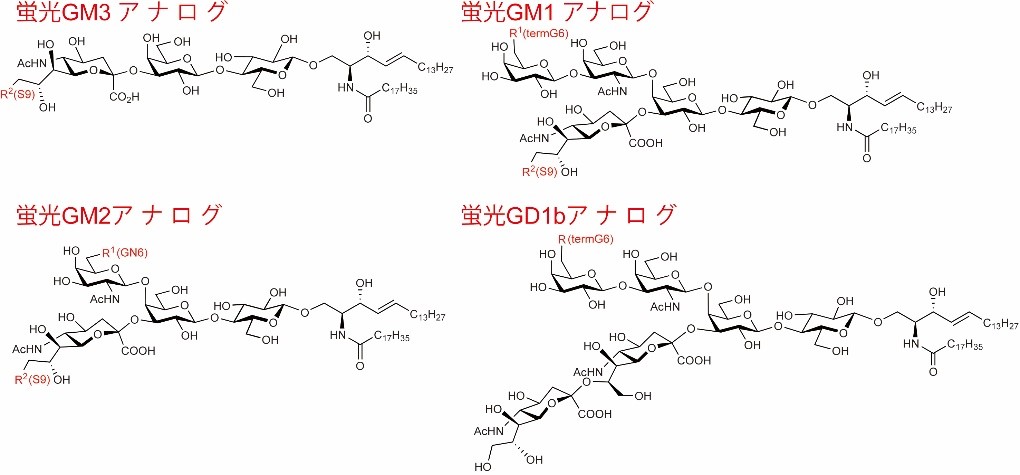
図6.人工膜系で、天然のガングリオシドと同様に挙動した蛍光標識ガングリオシドの構造
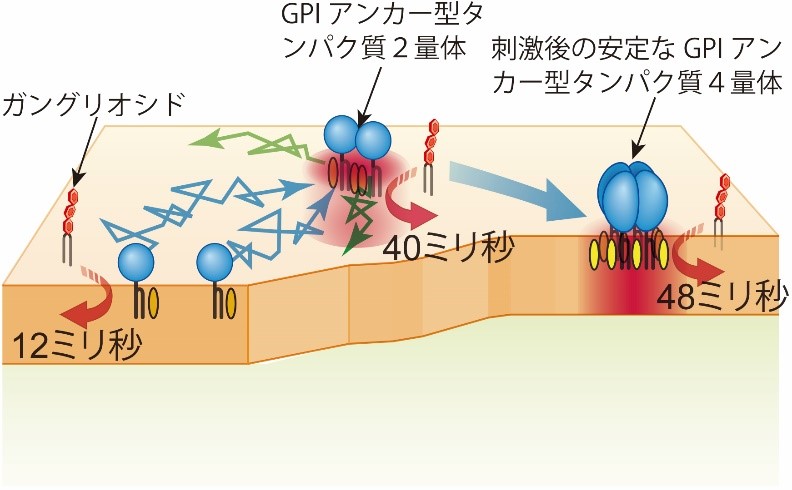
図7.ガングリオシドは、コレステロールがあるときにだけ、CD59単量体、二量体、そして安定化された四量体へ12~48ミリ秒間という短い間、リクルートされた。
プレスリリース2017 [Kinoshita and Suzuki et al., 2017 J. Cell Biol.]
6.ディジタル式シグナル伝達機構の解明
我々が今まで行ってきました、生細胞膜上の受容体とシグナル分子の2色同時1分子観察実験の結果や1分子FRET観察によりますと、シグナル分子は、わずか1秒以下の時間でリクルートされたり、活性化されていることが示唆されてきました(図4)。
一方で、western blottingなどにより観察される細胞全体のバルクシグナルの変化は、数分~20分程度続いていました。
これらの実験結果から、我々は「細胞膜上の受容体をリガンド刺激後、数分以上続く細胞全体のアナログシグナルは、1分子レベルで見ると1秒以下の時間しか続かないパルス状の短期間シグナルから積算されて作られる。」とするディジタル式シグナルシステム仮説を提案しました。
シグナルがディジタル式ですと、細胞全体のシグナル強度を変化させる際に、ただ単位時間あたりのパルスの数を変えて行けばいいので、複雑な制御を要せずノイズにも強いというメリットが考えられます。現在、この仮説を検証しています。
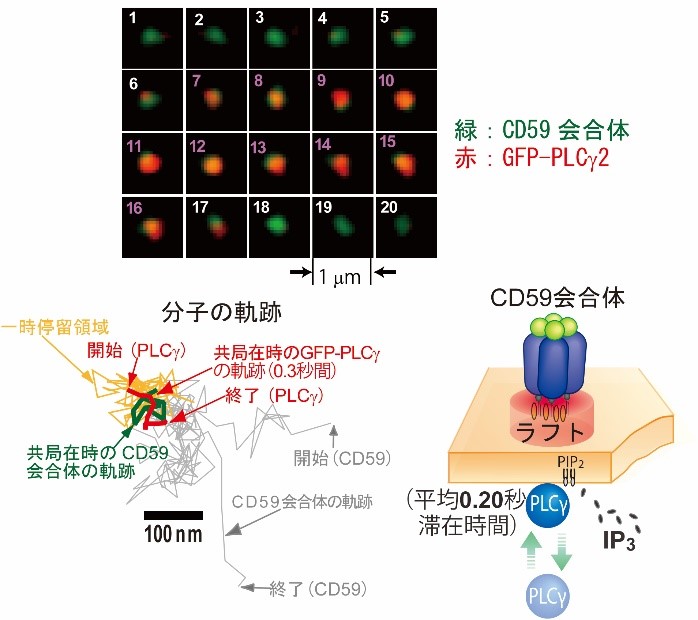
図8. PLCγのCD59会合体への短期間のリクルートの2色同時1分子観察の連続画像(上)、両者の軌跡(下左)と模式図(下右)。
[Suzuki et al., 2007a, J. Cell Biol.] [Suzuki et al. 2007b, J. Cell Biol.]
研究紹介
研究内容